Key Nodes and Representative Mechanisms in Angiogenic Regulatory Networks: A Structured Review
Key Nodes and Representative Mechanisms in Angiogenic Regulatory Networks: A Structured Review
Angiogenesis is a biological process in which new blood vessels arise from the pre-existing vasculature to maintain tissue perfusion and meet cellular demands for oxygen and nutrients. Its initiation and progression are finely orchestrated by multi-layer regulators, including soluble growth factors, cytokines, protease systems, extracellular matrix (ECM) remodeling, and cell-adhesion molecules. Dissecting the molecular circuitry of angiogenesis and the mechanisms underlying its pathological rewiring provides an essential theoretical foundation for mechanistic studies and targeted intervention in tumor progression, chronic inflammation, complications of metabolic diseases, and tissue repair.
Keywords: angiogenesis; exosomes; tumor microenvironment; inflammation and immunity; Wnt/β-catenin; hypoxia; extracellular matrix
I. Concept and Biological Significance of Angiogenesis
1.1 Core definition and major forms
Angiogenesis generally refers to the formation of new capillaries from an existing vascular network through sequential steps including endothelial activation, migration, proliferation, lumen formation, and maturation/stabilization. Two canonical modes are sprouting angiogenesis and intussusceptive angiogenesis, which can act in concert under distinct tissue microenvironments and mechanical contexts to drive vascular expansion and remodeling.
1.2 Boundaries between physiological and pathological angiogenesis
Physiological angiogenesis contributes to embryonic development, the menstrual cycle, and wound repair. In contrast, pathological angiogenesis is often characterized by abnormal structure and function (e.g., vascular malformations, increased permeability, reduced perfusion efficiency) and plays a pivotal role in tumor growth and metastasis, chronic inflammatory diseases, and impaired tissue repair in diabetes. A key distinction lies in whether the spatiotemporal scale of regulation and feedback closure is persistently amplified or becomes destabilized.
II. Key Molecules and Hierarchical Layers of Angiogenic Regulation
2.1 Soluble factor axes and receptor-mediated signaling
The VEGF family, FGF family, and Ang/Tie axis constitute core modules driving endothelial activation and the transition toward vessel stabilization. These factors form "parallel pathways" at the receptor level and converge on downstream cascades such as MAPK/ERK, PI3K/AKT, JAK/STAT, and SMAD to couple cell-cycle control, migratory cytoskeletal dynamics, and metabolic adaptation.
2.2 Extracellular matrix, protease systems, and adhesion networks
Protease systems, exemplified by matrix metalloproteinases (MMPs), degrade basement membranes and remodel the ECM to provide both physical space and biochemical cues for endothelial migration and lumenization. Integrins, selectins, and immune-cell adhesion molecules, in turn, determine the continuity of the "chemotaxis-adhesion-transmigration-colonization" event chain by shaping cell-matrix and cell-cell interactions.
2.3 Immune microenvironment and endothelial phenotypic reprogramming
Macrophages, fibroblasts, and tumor cells can jointly establish pro-angiogenic niches through secreted factors and vesicle-mediated communication. Immune infiltration not only supplies pro-angiogenic mediators, but can also alter endothelial responsiveness by modulating receptor expression programs and transcription-factor activity, thereby shifting a transient repair response toward sustained pathological neovascularization via increased feedback gain.
III. Structured Summaries of Representative Studies
3.1 Exosomal circCOL1A1 promotes colorectal cancer angiogenesis via the EIF4A3-Smad2/3 axis (reference [1])
【Research background and scientific question】
Angiogenesis is tightly linked to colorectal cancer progression, and the roles of circular RNAs in tumor communication and microenvironment remodeling have gained increasing attention. This study asked whether tumor-derived exosomes carrying circCOL1A1 reprogram endothelial angiogenic phenotypes via a defined RNA-binding protein and signaling pathway.
【Study design and key methods】
Along the "tumor cell-exosome-endothelial cell" axis, qRT-PCR, immunohistochemistry, and immunoblotting were used to quantify key molecules. HUVEC-based assays evaluated proliferation, migration, and tube formation. RIP, RNA pull-down, and FISH validated molecular interactions. Xenograft models provided in vivo validation.
【Key findings】
Exosomal circCOL1A1 enhanced angiogenic phenotypes in endothelial cells. EIF4A3 was upregulated in tumor tissues and contributed to endothelial responses. Smad2/3-related signaling was activated, and in vivo experiments showed accelerated tumor growth and angiogenesis.
【Mechanistic model and interpretation】
The pathway can be summarized as follows: tumor cells release exosomes enriched in circCOL1A1 → endothelial uptake enables circCOL1A1 to recruit EIF4A3 → EIF4A3 stabilizes Smad2/3-related mRNAs and amplifies pathway output → endothelial migration and tube-formation programs are induced.
【Translational implications and limitations】
This work supports exosomal circRNAs as upstream nodes in tumor angiogenic regulation, with potential utility as liquid biopsy markers or therapeutic targets. However, exosome uptake efficiency, in vivo distribution, and tissue specificity require quantitative assessment in models with closer clinical relevance.
3.2 The GRK2-PPARγ-Flt-1 axis regulates pro-angiogenic macrophage infiltration in rheumatoid arthritis (reference [2])
【Research background and scientific question】
In rheumatoid arthritis, synovial inflammation and aberrant angiogenesis reinforce each other. Although macrophage infiltration and polarization correlate with disease severity, the intracellular modules governing macrophage pro-angiogenic properties remained insufficiently resolved.
【Study design and key methods】
Using a collagen-induced arthritis model, myeloid-specific GRK2-deficient mice were evaluated for synovial inflammation and M1 polarization. RNA-seq and dual-luciferase reporter assays were used to identify interacting modules and downstream transcriptional axes. Pharmacological inhibition tested druggability.
【Key findings】
GRK2 deficiency alleviated synovial inflammation and M1 polarization. PPARγ was identified as a GRK2-associated module. The transcription of Flt-1 (VEGFR1), linked to migration and pro-angiogenic activity, was regulated by the GRK2-PPARγ axis. Inhibiting GRK2 activity reduced inflammation and angiogenic phenotypes associated with Flt-1+ macrophages.
【Mechanistic model and interpretation】
In inflammatory settings, increased GRK2 membrane recruitment → suppression of PPARγ activation and its transcriptional repression capacity → release of constraints on Flt-1 transcription → enhanced infiltration and pro-angiogenic effects of Flt-1+ macrophages → aggravated synovial inflammation and angiogenesis.
【Translational implications and limitations】
By mapping "immune-cell infiltration-angiogenesis" coupling to a pharmacologically actionable intracellular axis, the study provides a more targetable framework for intervening in angiogenesis linked to inflammatory diseases. Nevertheless, heterogeneity across joint microenvironments and macrophage subpopulations may influence translational generalizability.
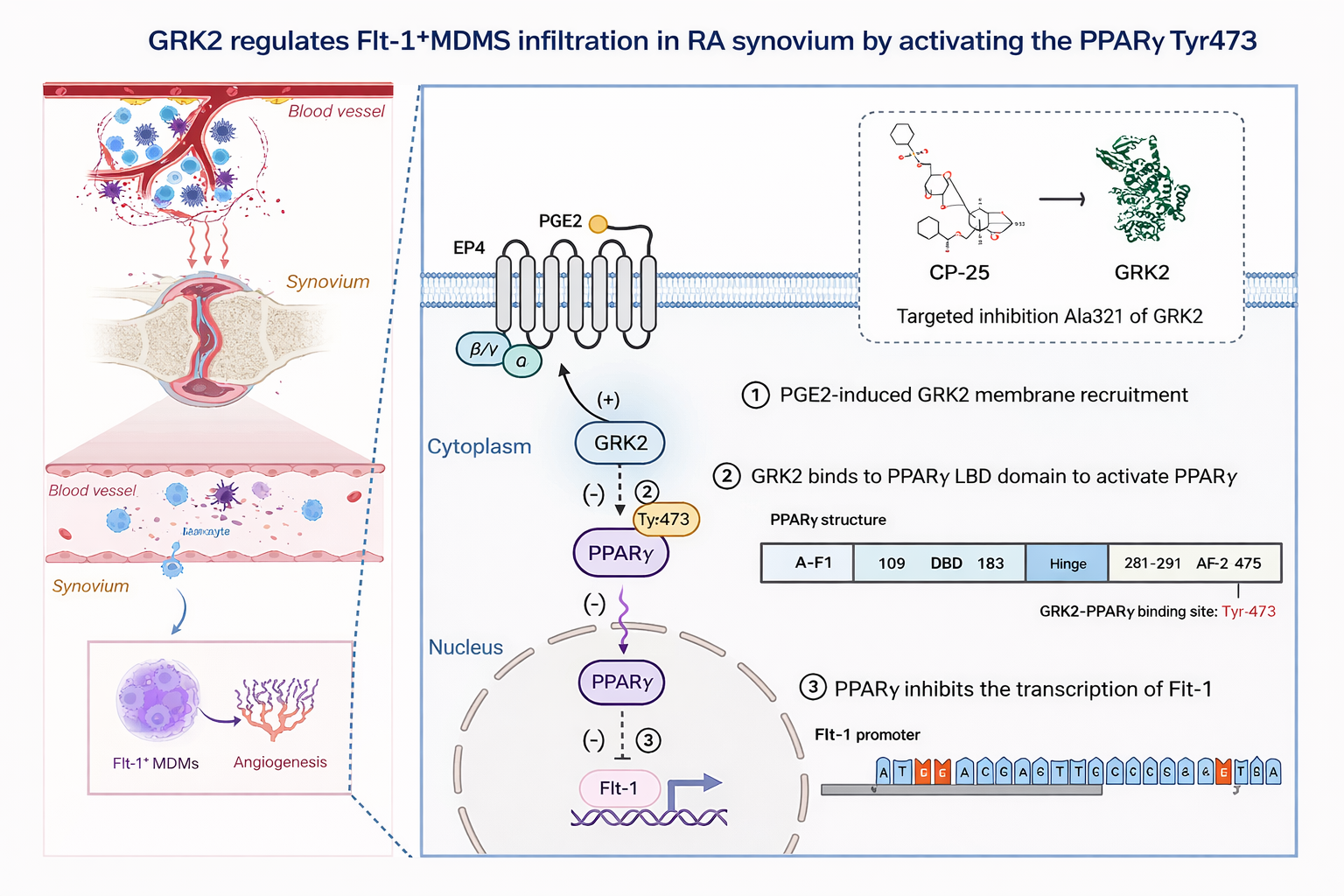
Fig1.GRK2 Activates PPARγ (Tyr473) to Suppress Flt-1 Transcription and Regulate Flt-1⁺ MDMS Infiltration in RA Synovium
3.3 sEV-derived vWF drives a tumor-endothelial positive feedback loop promoting angiogenesis and metastasis in hepatocellular carcinoma (reference [3])
【Research background and scientific question】
Hepatocellular carcinoma (HCC) is highly vascular, and tumor-endothelial crosstalk can sustain pro-angiogenic microenvironments. This study investigated whether proteins delivered by circulating small extracellular vesicles (sEVs) function as key amplifiers of tumor-endothelial interactions.
【Study design and key methods】
Candidate molecules were identified by circulating sEV proteomics with stage-associated analyses. In vitro assays tested endothelial angiogenesis, tumor-endothelial adhesion, and induction of signaling factors. In vivo studies assessed vascular leakage and metastasis. Antibody blockade and receptor inhibitors evaluated whether the feedback loop could be interrupted.
【Key findings】
sEV-associated vWF increased with HCC stage. Circulating sEVs from late-stage patients enhanced angiogenesis, tumor-endothelial adhesion, pulmonary vascular leakage, and metastasis; anti-vWF antibodies markedly attenuated these effects. Endothelial VEGF-A and FGF2 were upregulated and contributed to signal amplification, while FGF2 further activated a positive feedback on the tumor side via FGFR4/ERK1.
【Mechanistic model and interpretation】
A "vesicle delivery-endothelial activation-feedback amplification" loop emerges: tumor sEVs deliver vWF → endothelial cells are reprogrammed to release pro-angiogenic factors (e.g., VEGF-A, FGF2) → FGF2 feeds back to stimulate tumor signaling → pro-angiogenic and metastatic capacity is further reinforced, forming a self-amplifying system.
【Translational implications and limitations】
sEV-vWF may serve as a progression-associated liquid biomarker candidate and a therapeutic entry point for disrupting tumor-endothelial communication. Although combined blockade improved response in models, clinical safety windows, immune-related adverse effects, and stratification criteria require systematic evaluation.
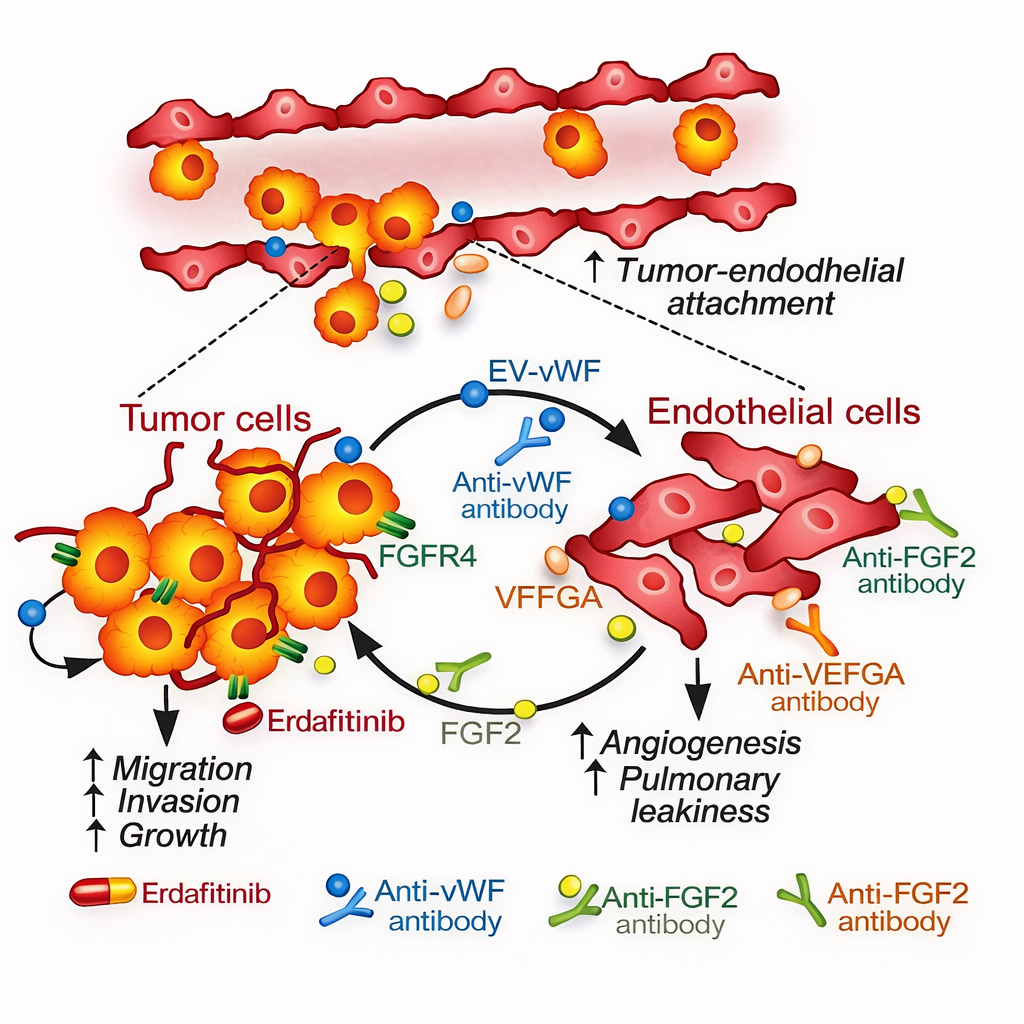
Fig2.Tumor Cell–Derived Extracellular Vesicles (EVs) Carrying vWF/FGF2/VEGFA Promote Tumor–Endothelial Adhesion and Increase Pulmonary Angiogenesis and Vascular Leakiness
3.4 Inhibiting cytosolic CXXC5 accelerates diabetic wound healing by enhancing angiogenesis and skin repair (reference [4])
【Research background and scientific question】
Chronic diabetic wounds (e.g., diabetic foot ulcers) are associated with inadequate angiogenesis, persistent inflammation, and reduced responsiveness of regenerative signaling. Wnt/β-catenin signaling is central to tissue regeneration but is often suppressed in diabetic wounds. This study proposed cytosolic CXXC5 as a negative regulatory node of Wnt/β-catenin and a potential target to restore angiogenesis and repair.
【Study design and key methods】
CXXC5 and Wnt/β-catenin-associated molecules were assessed in patient tissues and diabetic mouse wounds. The small molecule KY19334 was used to disrupt the CXXC5-Dvl interaction and restore pathway activity. Angiogenesis and repair outcomes were evaluated in wound healing and hindlimb ischemia models.
【Key findings】
Diabetic wound tissues displayed elevated CXXC5 with suppressed Wnt/β-catenin signaling. KY19334 markedly accelerated wound closure, enhanced angiogenesis, and promoted reconstruction of skin repair-associated structures. It also induced angiogenesis and improved perfusion in ischemia models.
【Mechanistic model and interpretation】
Diabetic milieu → elevated cytosolic CXXC5 inhibits Dvl-mediated Wnt/β-catenin transmission → regenerative and angiogenic gene programs become hypo-responsive → delayed healing. Disrupting the CXXC5-Dvl interaction removes this negative constraint → restores β-catenin transcriptional output → concurrently improves angiogenesis, re-epithelialization, and matrix remodeling.
【Translational implications and limitations】
This work supports a "regenerative pathway reset" paradigm for chronic non-healing wounds. However, efficacy robustness across microbial contexts, inflammatory phases, and concomitant medications, as well as the long-term safety boundaries of Wnt activation, warrant stringent translational studies.
3.5 Hypoxia-induced SUMOylation of hnRNP A2/B1 drives exosomal sorting of miR-204-3p and promotes glioblastoma angiogenesis (reference [5])
【Research background and scientific question】
Glioblastoma often resides in hypoxic microenvironments. Hypoxia reshapes not only proliferation and therapy tolerance, but also immune and vascular niches via exosomes. This study focused on how hypoxia regulates miRNA exosomal sorting and how this impacts endothelial angiogenesis.
【Study design and key methods】
Sequencing clues from patient cerebrospinal fluid/tissue were combined with cell models to validate the sorting trend of miR-204-3p. Molecular interaction and domain analyses identified the recognition basis of hnRNP A2/B1 in miRNA sorting. The roles of hypoxia and SUMOylation in hnRNP A2/B1 nucleocytoplasmic translocation and exosomal loading were assessed. Pathway interrogation and pharmacologic inhibition tested intervention feasibility.
【Key findings】
miR-204-3p exhibited enrichment in exosomes. Hypoxia increased SUMOylation of hnRNP A2/B1, promoting cytoplasmic translocation and thereby enhancing exosomal sorting of miR-204-3p. Exosomal miR-204-3p promoted endothelial tube formation via an ATXN1/STAT3-associated axis. SUMOylation inhibitors reduced sorting and suppressed tumor growth and angiogenesis-related phenotypes.
【Mechanistic model and interpretation】
Hypoxia → increased SUMOylation modifies hnRNP A2/B1 → hnRNP A2/B1 translocates from nucleus to cytoplasm → selective miRNA (miR-204-3p) is preferentially loaded into exosomes → exosomes deliver miR-204-3p to endothelial cells to activate pro-angiogenic signaling → a hypoxia-adaptive "sorting-delivery-reprogramming" pathway is established.
【Translational implications and limitations】
By connecting hypoxic adaptation to exosomal sorting via a druggable post-translational modification node (SUMOylation), this study provides a candidate entry point for targeting glioma-associated angiogenesis. However, potential context-dependent bidirectional effects of miR-204-3p across cell types and the systemic safety of SUMOylation inhibition must be carefully delineated.
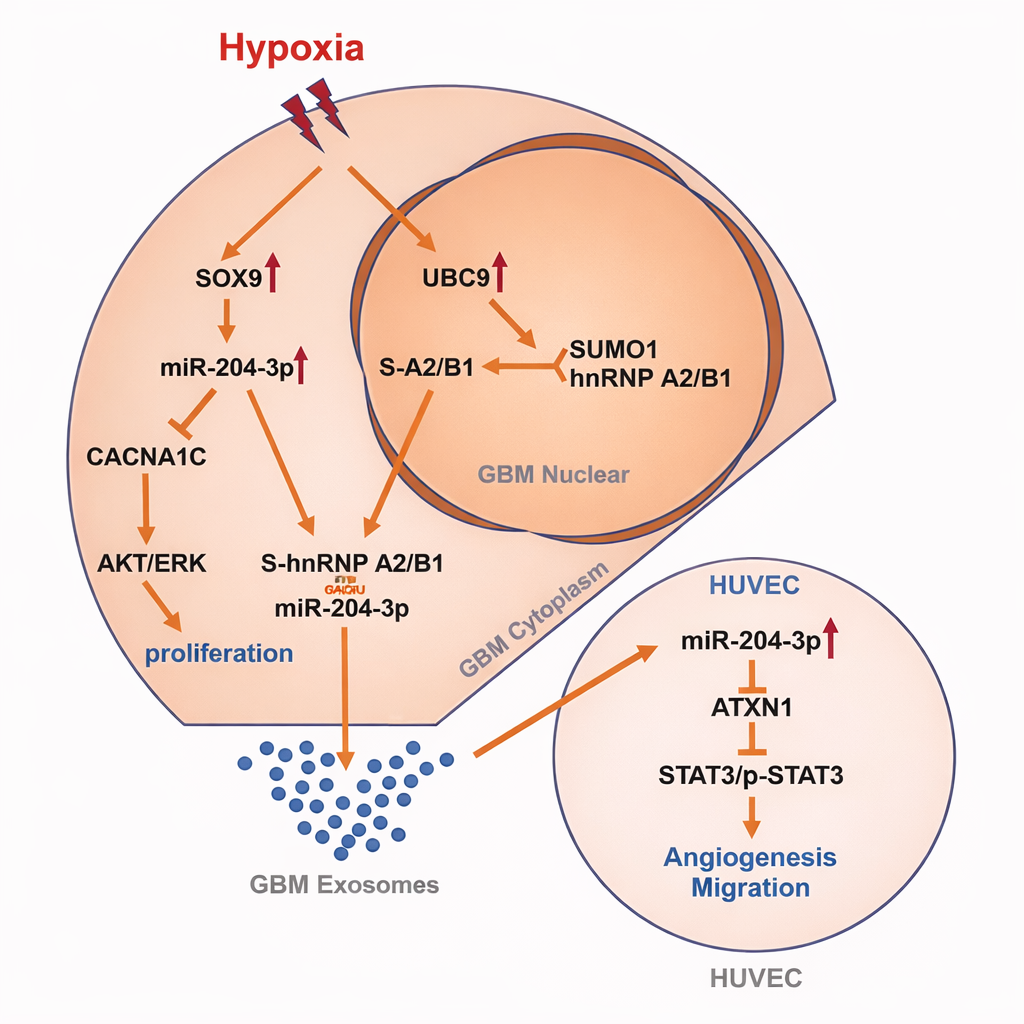
Fig3.Hypoxia-Induced SUMOylation of hnRNP A2/B1 in GBM Drives Proliferation and Angiogenesis via Exosomal miR-204-3p
IV. Cross-Study Commonalities and Experimental Strategy Considerations
4.1 Minimum closed-loop evidence chain
High-confidence mechanistic angiogenesis studies typically require a closed loop of evidence: clinical relevance, in vitro causality, and in vivo reproducibility. Clinical relevance anchors disease context and stratification signals; in vitro causality identifies key nodes and directionality; in vivo validation tests robustness within complex microenvironments and establishes intervention feasibility.
4.2 Multi-dimensional readouts for "pro-angiogenic" phenotypes
A single tube-formation assay cannot cover the full angiogenic program. It is preferable to combine readouts spanning endothelial proliferation/migration, matrix degradation, permeability, adhesion and transendothelial migration, perfusion function, and maturation/stabilization metrics, while explicitly mapping each readout to the corresponding biological stage to avoid stage-mismatch-driven interpretive bias.
4.3 Feedback amplification and tissue specificity as key determinants of translational difficulty
Positive feedback loops are common in tumors and chronic inflammation (e.g., vesicle-mediated communication that strengthens reciprocal interactions). These loops can yield high intervention benefit, yet are also prone to compensatory takeover by alternative pathways. Tissue specificity (e.g., liver, synovium, central nervous system) shapes immune-cell lineages, vascular architecture, and drug accessibility; stratification and control strategies should be incorporated at the study-design stage.
V. Aladdin-Related Products
Product Category | Product Name | Catalog No. | Grade and Purity | Application Positioning / Intended Use |
Antibody | Ramucirumab (anti-VEGFR2) | Carrier Free, Recombinant, ExactAb™, Azide Free, Validated, Animal Free, ≥95%(SDS-PAGE&SEC-HPLC), See COA | Blocks VEGFR2 signaling; inhibits endothelial proliferation and migration; functional control for the VEGF–VEGFR axis | |
Antibody | Ranibizumab (anti-VEGFA) | Carrier Free, Recombinant, ExactAb™, Low Endotoxin, Azide Free, Validated, Animal Free, ≥95%(SDS-PAGE&SEC-HPLC), See COA | Neutralizes VEGFA; assesses VEGFA-dependent angiogenesis; pathway-blocking control | |
Antibody | Recombinant VEGF Receptor 1 Antibody | See COA | VEGFR1 expression detection and mechanistic validation; suitable for receptor blockade or immunoassay workflows | |
Antibody | Recombinant VEGF Receptor 1 Antibody | ExactAb™, Validated, Recombinant, 0.1 mg/mL | VEGFR1 expression detection and mechanistic validation; suitable for receptor blockade or immunoassay workflows | |
Antibody | Recombinant VEGF Receptor 2 Antibody | Recombinant, ExactAb™, Validated, See COA | VEGFR2 expression detection and pathway validation; supports receptor-centric mechanistic studies | |
Antibody | Recombinant VEGF Receptor 2 Antibody | ExactAb™, Validated, Recombinant, 0.12 mg/mL | VEGFR2 expression detection and pathway validation; supports receptor-centric mechanistic studies | |
Antibody | Recombinant VEGFA Antibody | ExactAb™, Validated, Carrier Free, Recombinant, 0.075 mg/mL | VEGFA detection and neutralization/competition assays; angiogenic factor quantification and mechanistic validation | |
Antibody | Recombinant VEGFD Antibody | ExactAb™, Validated, Recombinant, 1.5 mg/mL | VEGFD detection and mechanistic studies; suitable for lymphangiogenesis or VEGFR3-related research workflows | |
Recombinant Protein | Recombinant Human VEGF 165 GMP Protein | ActiBioPure™, Bioactive, GMP, Animal Free, Carrier Free, High performance, ≥97%(SDS-PAGE&SEC-HPLC) | Canonical pro-angiogenic stimulus; establishes endothelial proliferation/migration and tube-formation models | |
Recombinant Protein | Recombinant Human VEGF 165 Protein | ActiBioPure™, GMP, Animal Free, Carrier Free, Bioactive, High Performance, sterile, ≥98%(SDS-PAGE) | Canonical pro-angiogenic stimulus; endothelial functional assays and efficacy benchmarking control | |
Recombinant Protein | Recombinant Human VEGF Protein | GMP, Bioactive, ActiBioPure™, High performance, Animal Free, Carrier Free, ≥95%(SDS-PAGE) | Pro-angiogenic stimulation; positive control for VEGF-axis assays and dose-window exploration | |
Recombinant Protein | Recombinant Human VEGFA-165 Protein | Animal Free, Carrier Free, Bioactive, ActiBioPure™, High Performance, See COA | Pro-angiogenic stimulation; supports endothelial signaling readouts and phenotype induction | |
Recombinant Protein | Recombinant Human VEGFR2/KDR Protein | Animal Free, Carrier Free, ActiBioPure™, Bioactive, High performance, ≥95%(SDS-PAGE) | Receptor-binding validation; ligand–receptor interaction studies and competitive inhibition assays | |
Recombinant Protein | Recombinant Human VEGFR3/Flt-4 Protein | Animal Free, Carrier Free, ≥95%(SDS-PAGE) | VEGFR3-related binding/blocking validation; supports lymphangiogenesis and receptor-mechanism studies | |
Recombinant Protein | Recombinant Mouse VEGF 164 Protein | ActiBioPure™, Bioactive, Animal Free, Carrier Free, Azide Free, High performance, ≥95%(SDS-PAGE) | Pro-angiogenic stimulation in murine systems; supports animal-derived cell models and in vitro functional assays | |
Recombinant Protein | Recombinant Rat VEGF 164 Protein | ActiBioPure™, Recombinant, Animal Free, Carrier Free, High performance, ≥95%(SDS-PAGE) | Pro-angiogenic stimulation in rat systems; suitable for rat-derived cells or comparative control experiments | |
Antibody | Recombinant FGF2 Antibody | Recombinant, ExactAb™, Validated, See COA | FGF2 detection/blockade and mechanistic validation; supports FGF-axis functional assays | |
Antibody | Recombinant FGF2 Antibody | ExactAb™, Validated, Recombinant, High performance, 0.103 mg/mL | FGF2 detection/blockade and mechanistic validation; supports FGF-axis functional assays | |
Recombinant Protein | Recombinant Human FGF basic/FGF2 Protein | ActiBioPure™, Carrier Free, High Performance, Bioactive, ≥90%(SDS-PAGE), See COA | Pro-angiogenic stimulus; acts in parallel with VEGF to drive endothelial proliferation and migration | |
Recombinant Protein | Recombinant Human FGF basic/FGF2/bFGF GMP Protein | Animal Free, Carrier Free, GMP, Bioactive, ActiBioPure™, High performance, ≥97%(SDS-PAGE&SEC-HPLC) | High-consistency FGF2 stimulation; suitable for organoid/stem cell culture and angiogenesis models | |
Recombinant Protein | Recombinant Human FGF basic/FGF2/bFGF Protein | Carrier Free, GMP, Bioactive, ActiBioPure™, High Performance, Animal Free, ≥95%(SDS-PAGE) | FGF2 stimulus; supports endothelial functional assays and efficacy benchmarking controls | |
Recombinant Protein | Recombinant Human FGF basic/FGF2/bFGF(155aa) Protein | GMP, Bioactive, ActiBioPure™, Animal Free, Carrier Free, High Performance, ≥95%(SDS-PAGE) | FGF2 stimulus; supports dose–response studies and signaling readout validation | |
Recombinant Protein | Recombinant Mouse FGF basic/FGF2/bFGF Protein | Animal Free, Carrier Free, Azide Free, ≥98%(SDS-PAGE) | FGF2 stimulation in murine systems; supports animal-derived endothelial/fibroblast models | |
Antibody | Recombinant Angiopoietin 1 Antibody | Recombinant, ExactAb™, Validated, See COA | Ang/Tie-axis detection and mechanistic validation; supports studies of vascular maturation and stabilization | |
Antibody | Recombinant Angiopoietin 2/ANG2 Antibody | ExactAb™, Validated, Recombinant, High performance, 0.7 mg/mL | ANG2 mechanistic studies; supports vascular destabilization and inflammation-associated angiogenesis models | |
Antibody | Recombinant Angiopoietin-like 4/ANGPTL4 Antibody | ExactAb™, Validated, Recombinant, 0.8 mg/mL | ANGPTL4 detection and mechanistic validation; supports studies of vascular permeability and tumor microenvironment biology | |
Recombinant Protein | Recombinant Human Angiopoietin-like 4/ANGPTL4 Protein | ActiBioPure™, Bioactive, Animal Free, Carrier Free, Azide Free, ≥95%(SDS-PAGE) | ANGPTL4 functional studies; evaluates effects on permeability, migration, and microenvironmental regulation | |
Recombinant Protein | Recombinant Human Angiopoietin-like 7 Protein | Animal Free, Carrier Free, ≥95%(SDS-PAGE), See COA | ANGPTL7 research reagent; functional validation in angiogenesis and tissue remodeling contexts | |
Recombinant Protein | Recombinant human ANGPTL7 protein | ActiBioPure™, Bioactive, Animal Free, Carrier Free, Azide Free, ≥95%(SDS-PAGE) | ANGPTL7 research reagent; supports mechanistic and phenotypic assays | |
Assay / Functional Measurement | FRETS-VWF73 | -- | Substrate for vWF cleavage functional assays; suitable for assessing vWF-related protease activity and mechanism studies | |
Recombinant Protein | Recombinant Human vWF-A2 Protein | Carrier Free, ≥95%(SDS-PAGE), See COA | vWF-A2 domain mechanistic studies; supports cleavage/binding assays to validate pathway steps | |
siRNA | VWF Human Pre-designed siRNA Set A | -- | vWF knockdown intervention; establishes causality of vWF in endothelial activation and angiogenic phenotypes | |
Antibody | PLGF Mouse mAb | Carrier Free, ExactAb™, Azide Free, Validated, See COA | PlGF detection and functional blockade; supports VEGFR1-related angiogenesis mechanism validation | |
Antibody | PLGF Mouse mAb | Carrier Free, ExactAb™, Azide Free, Validated, See COA | PlGF detection and functional blockade; supports VEGFR1-related angiogenesis mechanism validation | |
Recombinant Protein | PLGF-I human | Animal Free, Recombinant, for cell culture, ≥95%(SDS-PAGE&HPLC), expressed in E. coli | PlGF stimulus; supports VEGFR1-axis activation and phenotype induction | |
Recombinant Protein | Recombinant Human PLGF Protein | ActiBioPure™, Bioactive, Animal Free, Carrier Free, Azide Free, High performance, ≥97%(SDS-PAGE&HPLC) | PlGF stimulus; evaluates VEGFR1-dependent migration and angiogenic effects | |
Recombinant Protein | Recombinant Human PlGF Protein | ActiBioPure™, Bioactive, Animal Free, Carrier Free, Azide Free, ≥95%(SDS-PAGE) | PlGF stimulus; supports angiogenesis models and mechanistic validation workflows | |
Recombinant Protein | Recombinant Human PlGF Protein | Animal Free, Carrier Free, Bioactive, ActiBioPure™, ≥90%(SDS-PAGE), See COA | PlGF stimulus; used as a parallel pro-angiogenic factor or comparative stimulation control | |
Recombinant Protein | Recombinant Human SMAD2 Protein | Carrier Free, ≥90%(SDS-PAGE), See COA | Smad2/3-pathway in vitro mechanistic support; suitable for binding/activity assays or antibody-validation systems | |
Recombinant Protein | Recombinant Human Smad3 Protein | Carrier Free, Azide Free, ≥90%(SDS-PAGE) | Smad3 mechanistic studies; supports signaling-pathway and interaction validation | |
Recombinant Protein | Recombinant Human Smad3 Protein | Carrier Free, Azide Free, ≥95%(SDS-PAGE) | Smad3 mechanistic studies; supports signaling-pathway and interaction validation | |
Recombinant Protein | Recombinant Human SMAD4 Protein | Carrier Free, ≥90%(SDS-PAGE), See COA | Smad4 mechanistic studies; supports complex formation and downstream transcriptional-axis research | |
Antibody | Recombinant Smad2 Antibody | Recombinant, ExactAb™, Validated, See COA | Smad2 detection and pathway validation; suitable for Western blot and immunoassay readouts | |
Antibody | Recombinant SMAD3 Antibody | ExactAb™, Validated, Recombinant, 1 mg/mL | Smad3 detection and pathway validation; supports mechanistic attribution and intervention controls | |
Antibody | Recombinant Smad4 Antibody | ExactAb™, Validated, Recombinant, 0.6 mg/mL | Smad4 detection and pathway validation; supports mechanistic attribution and intervention controls | |
Recombinant Protein | Recombinant Human STAT3 Protein | Carrier Free, ≥90%(SDS-PAGE), See COA | STAT3-axis mechanistic support; suitable for binding/activity assays or antibody-validation systems | |
Antibody | Recombinant STAT3 Antibody | Recombinant, ExactAb™, KD Validation, Validated, See COA | STAT3 detection and pathway validation; supports angiogenesis-associated inflammatory signaling readouts | |
Small-Molecule Inhibitor | GM 6001 | Moligand™, ≥98% | Inhibits MMP-mediated matrix degradation; inhibitory control for migration, invasion, and angiogenic phenotypes | |
Small-Molecule Inhibitor | MMP Inhibitor II | ≥95% | Broad-spectrum MMP inhibition; validates the contribution of ECM remodeling to angiogenesis and migration | |
Small-Molecule Inhibitor | NNGH | ≥98% | MMP inhibition; supports matrix-degradation–dependent mechanistic validation | |
Small-Molecule Inhibitor | NSC 405020 | ≥98% | Inhibits MT1-MMP–associated functions; validates endothelial invasion and matrix-transmigration steps | |
Small-Molecule Inhibitor | PD 166793 | ≥99%(HPLC) | MMP inhibition; pharmacological control for angiogenesis and migration phenotypes | |
Small-Molecule Inhibitor | TAPI 0 | ≥95%(HPLC) | Inhibits ADAM17-mediated shedding and MMP activity; supports inflammation–angiogenesis coupling mechanisms | |
Small-Molecule Inhibitor | TAPI 2 | ≥95%(HPLC) | Inhibits ADAM17-mediated shedding and MMP activity; supports receptor/ligand shedding–dependent mechanistic studies | |
Small-Molecule Inhibitor | S3I-201 | ≥96% | STAT3 pathway inhibition; mechanistic attribution for pro-angiogenic inflammatory signaling and transcriptional outputs | |
Small-Molecule Inhibitor | STA-21 (Ochromycinone) | ≥98% | STAT3 inhibition control; suppresses pro-angiogenic gene programs | |
Small-Molecule Inhibitor | WP1066 | Moligand™, ≥98% | Inhibits the JAK/STAT3 axis; validates cytokine-driven angiogenic signaling dependency | |
Small-Molecule Inhibitor | AMG PERK 44 | ≥95% | Inhibits the ER stress–PERK pathway; control for probing angiogenic adaptive mechanisms under hypoxia/stress conditions | |
Small-Molecule Inhibitor | GSK PERK inhibitor | Moligand™, ≥98% | PERK pathway inhibition; validates stress-associated angiogenic phenotypes | |
Small-Molecule Inhibitor | GSK2606414 | Moligand™, ≥98% | PERK inhibition; validates stress-driven transcriptional reprogramming coupled to angiogenesis | |
Small-Molecule Inhibitor | GSK2656157 | Moligand™, ≥98% | PERK inhibition; pathway-blocking control for stress-associated signaling | |
Small-Molecule Inhibitor | ERK inhibitor | ≥95% | Inhibits MAPK/ERK output; validates downstream pathway dependency after VEGF/FGF stimulation | |
Small-Molecule Inhibitor | ERK Inhibitor III | Moligand™ | ERK inhibition control; supports rapid blockade of MAPK/ERK signaling and phenotype-reversal validation | |
Small-Molecule Inhibitor | ERK Inhibitor II, Negative Control | Moligand™ | Negative control; excludes non-specific effects and calibrates interpretation of ERK inhibition experiments | |
Small-Molecule Inhibitor | FR 180204 | Moligand™, ≥98%(HPLC) | ERK inhibition; validates ERK dependency of endothelial proliferation, migration, and tube formation | |
Small-Molecule Inhibitor | Terrein | ≥98% | Dual inhibition of AKT and ERK; supports parallel-pathway blockade and attribution of pro-angiogenic phenotypes | |
Small-Molecule Inhibitor | AZD4547 | Moligand™, ≥99% | FGFR inhibition; validates FGF/FGFR-driven angiogenesis and migration mechanisms | |
Small-Molecule Inhibitor | BGJ398 (NVP-BGJ398) | Moligand™, ≥98% | FGFR inhibition; supports FGF-axis dependency validation and efficacy controls | |
Small-Molecule Inhibitor | CH5183284 (Debio-1347) | Moligand™, ≥99% | FGFR inhibition; blocks FGFR-dependent angiogenic signaling | |
Small-Molecule Inhibitor | LY2874455 | Moligand™, ≥99% | Broad FGFR inhibition; supports phenotype-reversal validation for FGF-axis involvement | |
Small-Molecule Inhibitor | PD-161570 | ≥98%(HPLC) | FGFR inhibition; validates receptor-kinase dependency and explores dose windows | |
Small-Molecule Inhibitor | SSR128129E | ≥98% | Allosteric FGFR inhibition; supports FGF signaling blockade and pathway-specificity validation | |
Small-Molecule Inhibitor | AAL 993 | ≥98% | VEGFR pathway inhibition; validates angiogenesis dependency and efficacy controls | |
Small-Molecule Inhibitor | AEE788 (NVP-AEE788) | ≥97% | Dual inhibition of EGFR and VEGFR; supports pathway crosstalk and compensatory-mechanism studies |
Angiogenesis is not a linear process driven by a single pathway; rather, it is a dynamic network co-woven by soluble factors, ECM remodeling, immune microenvironments, and vesicle-mediated communication. The five representative studies, respectively centered on exosomal circRNA, immune-cell intracellular signaling axes, sEV protein-driven positive feedback loops, regenerative pathway resetting, and hypoxia-linked post-translational modification, illustrate diverse mechanistic patterns by which pathological angiogenesis is persistently amplified or, conversely, re-aligned toward repair-associated reconstruction. For translation-oriented research, priorities include: locking druggable nodes through rigorous evidence closure, defining benefit windows via refined population and disease-stage stratification, and addressing feedback compensation and tissue specificity through a systems-biology perspective.
References
[1] Hu G, Lin C, Gao K, Chen M, Long F, Tian B. Exosomal circCOL1A1 promotes angiogenesis via recruiting EIF4A3 protein and activating Smad2/3 pathway in colorectal cancer. Molecular Medicine. 2023;29:155. doi:10.1186/s10020-023-00747-x.
[2] Yang X, Zhao Y, Wei Q, Zhu X, Wang L, Zhang W, et al. GRK2 inhibits Flt-1+ macrophage infiltration and its proangiogenic properties in rheumatoid arthritis. Acta Pharmaceutica Sinica B. 2024;14(1):241-255. doi:10.1016/j.apsb.2023.09.013.
[3] Wong SWK, Tey SK, Mao X, Fung HL, Xiao ZJ, Wong DKH, et al. Small Extracellular Vesicle-Derived vWF Induces a Positive Feedback Loop between Tumor and Endothelial Cells to Promote Angiogenesis and Metastasis in Hepatocellular Carcinoma. Advanced Science. 2023;10(26):2302677. doi:10.1002/advs.202302677.
[4] Kim E, Seo SH, Hwang Y, Ryu YC, Kim H, Lee KM, et al. Inhibiting the cytosolic function of CXXC5 accelerates diabetic wound healing by enhancing angiogenesis and skin repair. Experimental & Molecular Medicine. 2023;55(8):1770-1782. doi:10.1038/s12276-023-01064-3.
[5] Guo Q, Fan Y, Wang Q, Li B, Qiu W, Qi Y, et al. Glioblastoma upregulates SUMOylation of hnRNP A2/B1 to eliminate the tumor suppressor miR-204-3p, accelerating angiogenesis under hypoxia. Cell Death & Disease. 2023;14:147. doi:10.1038/s41419-023-05663-w.
For more related articles, please see below:
[1] Tumor cell-induced angiogenesis model experiment
Aladdin: https://www.aladdinsci.com/