Mammalian protein-protein interaction traps based on fluorescence-activated cell sorting technology
Mammalian protein-protein interaction traps based on fluorescence-activated cell sorting technology
The Mammalian Protein-Protein Interaction Trap System (MAPPIT) is a two-hybrid system designed based on the understanding of the signaling mechanism of type I cytokine receptors. 'Decoy' peptides and 'prey' peptides are linked to mutant cytokine receptor chimeras, which are impaired in signaling. Upon stimulation with the appropriate ligand, if the 'bait' and 'prey' can interact and bind, the JAK-STAT signaling cascade amplification system is initiated, inducing a large number of transcripts of reporter genes or marker genes controlled under the STAT3-responsive rPAP1 (rat pancreatitis associat-ed protein 1) promoter. In addition to providing a relevant physiological context for mammalian protein-protein interaction systems, this approach also provides segregated action and effector regions that can be used for analytical and screening purposes.
Principle
The basic principle of the Mammalian Protein-Protein Interaction Trap System (MAPPIT) is that a "bait" peptide and a "prey" peptide are linked to mutant cytokine receptor chimeras whose signaling is impaired. Upon stimulation with the appropriate ligand, if the 'bait' and 'prey' can interact and bind, the JAK-STAT signaling cascade amplification system is activated, inducing massive transcription of reporter or marker genes under the STAT3-responsive rPAP1 (rat pancreatitis associat-ed protein 1) promoter, which provides segregated action and effector regions, which in combination with flow cytometry can be used to analyze and screen cells.
Operation method
Establishment of a mammalian protein-protein interaction trap system based on fluorescence-activated cell sorting technology
Principle
The basic principle of the Mammalian Protein-Protein Interaction Trap System (MAPPIT) is that a "bait" peptide and a "prey" peptide are linked to a mutant cytokine receptor chimera whose signaling is impaired. Upon stimulation with the appropriate ligand, if the "bait" and "prey" are able to interact and bind, the JAK-STAT signaling cascade amplification system is activated, inducing the activation of an associat-ed protein 1 (rPAP1) controlled by STAT3 in response to rPAP1 (rat pancreatitis). associat-ed protein 1 (rPAP1) promoter, this approach provides segregated regions of action and effectors, which can be used in conjunction with flow cytometry to analyze and screen cells.
Materials and Instruments
Equipment: Move The basic process of establishing a mammalian protein-protein interaction trap system based on fluorescence-activated cell sorting technology can be divided into the following steps: 1.1 Cell line modification for cDNA library screening by FACS method Human IL-5 receptor α subunit was used, and in the established expression complex, hIL-5Rα surface tag was controlled by the STAT3-dependent rPAP1 promoter. After co-transfection of HEK 293 Flp-In T-REx cells, clones that stably integrated the expression complex with the EcoR expression body were selected. After stimulation with leukemia inhibitory factor (signaling via endogenous expression receptor via STAT3 pathway), hIL-5Ra levels were measured by flow cytometry. In the assay, α16 and AlexaFluor488 labeled anti-mouse IgG were used as primary and secondary antibodies, respectively. 1.2 Expression vectors for receptor-decoy chimeras The pCELIf and pCLL1f vectors used for the expression of the receptor-decoy chimera were derived from the peDNA5/FRT plasmid (Flp-In system), which contains the FRT locus and is linked to the thaumatin resistance gene, which is capable of integrating into T-Rex44 host cells for the sake of the Flp recombinase and screening for stabilized cell lines. Fusion receptor expression is controlled by the human cytomegalovirus promoter. pCEL1f and pCLL1f plasmids differ in the nature of the extracellular region of the chimeric receptor, which is derived from the human erythropoietin receptor (EpoR) or the murine leptin receptor (LR)). Both chimeric receptors were further linked to the transmembrane and intracellular portions of the leptin receptor variant, which has its tyrosine replaced with phenylalanine (LR-F3). The decoy protein (fragment) was cloned after a short hinge sequence using SacI and Notl restriction enzyme cleavage sites. Upon co-transfection of the decoy vector and the Flp recombinase expression plasmid POG44 (Invitrogen), recombination between the FRT site of the decoy vector and the host cell genome resulted in the conversion of bleomycin resistance to chaotropic acid resistance (by inducing a polyadenylate signal sequence in front of the LacZ-Zeocin fusion gene) and stable expression of the receptor-decoy chimera. (B) Sketch of the pBG1 prey vector. This vector includes the E.coli ccdB reverse selection complex for easy cDNA library proliferation. 1.3 Prey cDNA Library The prey cDNA library was obtained from pBG1, which is derived from pBABE, a murine retroviral vector containing the Molo-ney leukemia virus long terminal repeat and ψ sequences. pBG1 plasmid encodes a FLAG-tagged gp130 fragment, which has a GGS (Gan-Gan-Silk) amino acid linkage region followed by a polyclonal site that is cleaved by EcoRI and Notl or Xhol. This is followed by multiple cloning sites into which the cDNA can be unidirectionally cloned using EcoRI and Notl or Xhol cleavage sites. To enable reverse selection of self-associating plasmids, the integrated prey sequence removes the filler sequence encoding the bacterial ccdB protein (control of cell death B). libraries of Oligo(dT) or randomly primed cDNAs, or a combination of both, can be used for screening.
① FACS sorting tube (Falcon).
② FACSVantage SE flow cytometer (BD Biosciences, San Jose, CA) (see Subheading 3.3.3.).
③ FlowJo analysis software (Tree Star, Inc., San Carlos, CA) or other flow cytometry analysis software.
④ Centrifuge
Reagents:
① Flp-In T-REx system (Invitrogen, Carlsbad, CA) or T-Rex44 cell line.
② Plat-E cell line.
③ pXP2d2-rPAP1-hIL-5RαΔcyt.
④ pCEL1f, pCLL1f (derived from pcDN A5/FRT vector; Flp-In system, Invitrogen) and PBG1 vector (derived from PBABE vector).
⑤ Culture medium: DMEM containing 10% fetal bovine serum.
⑥ Human leukemia inhibitory factor (LIF) (Sigma-Aldnich, St. Louis, MO), human erythropoietin (Epo) (R & D Sgstms, Abingdon, Oxfordshire, UK) and murine leptin (R & D Systems).
(vii) Hair laryngeal pigment Forskolin, (Sigma-Aldrich).
⑧ Cell dissociation reagent (Invitrogen), penicillin/streptomycin (Invitrogen).
⑨ Polybrene (Polybrene; Sigma-Aldrich).
⑩ Hygromycin B (Invitrogen) and Zeocin (Invitrogen).
⑪ Mouse α16 monoclonal antibody against human IL-5Rα extracellular region.
⑫ Antibodies against the extracellular region of the human Epo receptor (e.g., Sheep Polyantibody, Catalog No. AF-322-PB, R&D Systems) and the extracellular portion of the murine leptin receptor.
⑬ Fluorescein-labeled secondary antibodies (e.g., Alexa Flaor 488 coupled antibody, Molecular Probes, Eugene, OR).
⑭ Magnetic bead sorting kit reagents (Miltenyi Biotec, Begisch-Gladbach, Germany Mid: MACS device, LS column, anti-biotin-coupled MACS microspheres).
⑮ FACS buffer: no Ca2
+
and Mg2
+
PBS without Ca2+and Mg2+ , containing 1% fetal bovine serum, 0.5 mM EDTA and 0.1% gentamicin. 0.22 μm filtration sterilized and stored at 4 °C. ⑯ FACS buffer.
⑯ Propidium iodide (P1).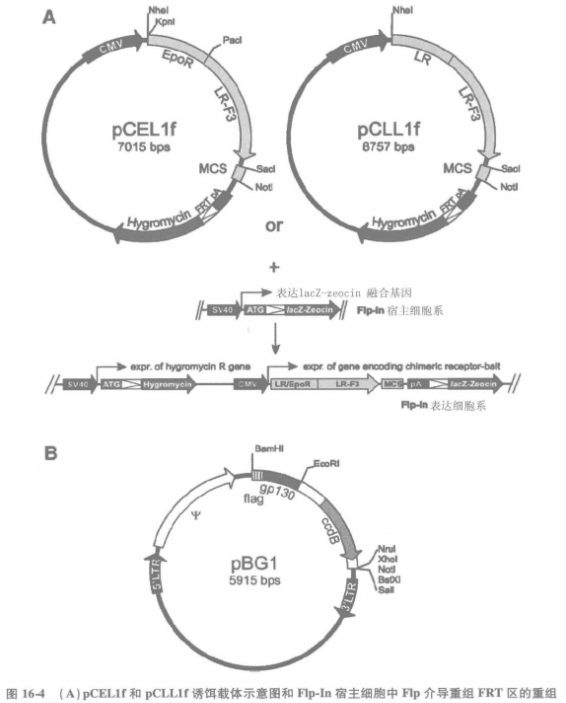
2.1 Establishment of a pool of homozygous cells stably expressing receptor-decoy fusions
Receptor-decoy chimeric expression vectors were transfected into T-Rex44 cells and a pool of homozygous cells stably expressing the fusion proteins was screened according to the instructions. Cell membrane expression of the receptor-decoy protein chimeras was confirmed by FACS analysis.
2.1.1 4x105 homozygous cells were inoculated in 10 cm2 tissue culture plates with 2 ml of culture medium (including negative control parental FRex 44 cells) at 37 ℃, 8%~10% CO2 overnight.
2.1.2 Separate the cells with 500 μl of cell digest.
2.1.3 Remove the digest, add 500 μl of FACS buffer for 2 min, and centrifuge at 150 g to collect the cells.
2.1.4 Resuspend the cells with 100 μl of FACS buffer and add antibodies against the extracellular portion of the receptor-decoy fusion (we use goat anti-human EpoR polyantibody at a final concentration of 2 μg/ml for PCELIf vector expression, and then homemade rat anti-mouse LR monoclonal antibody 4A9 or 1 G2 at a final concentration of 3 μg/ml for PCLLIf expression vectors). The antibody was mixed at 4 ℃ for 2 h on a rotary mixer.
2.1.5 Wash the cells carefully: add 900 μl of FACS buffer, centrifuge at 150 g for 2 min, then add 500 μl of FACS buffer, gently vortex and mix, then centrifuge at 150 g for 2 min.
2.1.6 Resuspend the cells with 100 μl of FACS buffer, add suitable fluorescein-coupled secondary antibody (we use Alexa Fluor488 coupling antibody), 4 ℃, 45 min.
2.1.7 Wash the cells with 900 μl of FACS buffer and centrifuge at 150 g for 2 min.
2.1.8 Resuspend cells in 250 μl of FACS buffer and add 3 μM propidium iodide.
2.1.9 FACS analysis to determine the expression of the receptor-decoy fusion.
2.2 Determination of screening conditions
To enable MACS pre-sorting, cells carrying the hIL-5Ra surface marker were first labeled with biotinylated α16 and then reacted with anti-biotin-antibody coupled MACS microspheres. Cells are separated by placing them in a separation column with a strong magnetic field, and the effluent is highly enriched for positive cells.The advantage of MACS pre-sorting is that it removes some of the prey non-dependently reduced hIL-5Rα expression. Different decoys may have different backgrounds and screening conditions with ideal signal-to-noise ratios should be determined for each decoy protein.
2.2.1 To test each screening condition, as well as negative and positive controls, 5 × 106 homozygous cells were seeded in 75 cm2 tissue culture flasks with 20 ml of culture medium and incubated overnight at 37 °C in 8%~10% CO2.
2.2.2 Add Epo or leptin according to the screening conditions of the test, and incubate at 37 ℃ with 8%~10% CO2 for 24 hours.
2.2.3 Digest cells with digestive solution, collect, and filter through a 70 μm cell filter.
2.2.4 Dilute the digest with FACS buffer and collect the cells by centrifugation.
2.2.5 Resuspend the cells with FACS buffer to a concentration of 10 cells/ml, add biotinylated α16 antibody (final concentration 1 μg/ml), and mix for 2 h at 4 ℃ on a rotary mixer.
2.2.6 Carefully wash the cells: add 10-20 times the volume of FACS buffer, 500 g, centrifuge for 3 min, resuspend the cells with FACS buffer, 500 g and centrifuge for 3 min.
2.2.7 For every 107 cells, resuspend the cells with 80 μl of FACS buffer, add 20 μl of anti-biotin antibody MACS microspheres, mix gently with a vortexer, and incubate for 15 min at 6~12 ℃.
2.2.8 Wash the cells: 10-20 times the volume of FACS buffer, 500 g, centrifuge for 3 min, resuspend the cells with FACS buffer, centrifuge again at 500 g for 3 min.
2.2.9 For every 10 cells, add 500 μl of de-airing FACS buffer to resuspend the cells, using a minimum of 1 ml.
2.2.10 Perform MACS sorting according to the manufacturer's instructions: place the LS column on the MACS sorting magnet and wash the column with 3 ml of FACS buffer.
2.2.11 After the cells have been filtered through a 70 μm cell strainer, add the cells to the MACS column and collect the effluent.
2.2.12 Wash the column three times with 3 ml FACS buffer. Collect the effluent as in the previous step (incorporate the elution fractions).
2.2.13 Remove the column from the separator, add 5 ml of de-aerated FACS buffer, and collect the cells in a collection tube by extruding them with a syringe (this is the portion of the sample that is sorted).
2.2.14 After centrifugation of the cells at 500 g for 3 min, resuspend the cells in FACS buffer, add the appropriate fluorescein-labeled secondary antibody (we commonly use AlexaFluar488 coupled anti-mouse IgG antibody, 10/cell with a final concentration of 1.3 μg/ml of secondary antibody) at 4 ℃, and mix for 45 min with a rotary mixer.
2.2.15 Add 10-20 times the volume of FACS buffer 500 g, 3 min, wash the cells.
2.2.16 Resuspend the cells with FACS buffer and add 3μM propidium iodide.
2.2.17 Analyze the background content of hIL-5Rα marker expression by FACS, and record the number of cells in the washed-out fraction and the sorted fraction, in order to analyze the recovery rate under different screening conditions.
3 Screening3.1 Retroviral infection and stimulation
3.1.1 Plant 10 homozygous cells into 175 cm2 tissue culture flasks, add 20 ml of medium, incubate for 5 h at 37 ℃, 8%~10% CO2.
3.1.2 Add polyglutamine at a final concentration of 2.5 μg/ml, and add viral particles containing cDNA libraries at appropriate dilution. At the same time, prepare at least one bottle of mock transfected cells as a control for FACS analysis.
3.1.3 Incubate at 37 ℃, 8%~10% CO2 overnight.
3.1.4 Change the culture medium and add Epo or leptin (with/or without trichostatin).
3.1.5 Incubate at 37℃, 8%~10% CO2 overnight.
3.2 MACS pre-sorting and FACS single-cell sorting
3.2.1 Prepare MACS sorted cells according to steps 3~10 of Subheading 2.2.
3.2.2 Place the LS column in the MACS separation magnet and wash the column with 3 ml of degassed FACS buffer.
3.2.3 Filter the cells through a 70 μm cell strainer and add (1~10 ml) to the separation column.
3.2.4 Wash the column 3 times with 3 ml of de-aerated FACS buffer.
3.2.5 Remove the column from the separator, add 5 ml of de-airing FACS buffer and squeeze the cells into a collection tube with a syringe.
3.2.6 After centrifugation of the cells at 500 g for 3 min, resuspend the cells in FACS buffer, add the appropriate fluorescein-labeled secondary antibody (our 10/ml cells with anti-mouse IgG antibody coupled to AlexaFluor488 at a final concentration of 1.3 μg/ml), and mix for 45 min at 4 °C on a rotary mixer.
3.2.7 Add 10-20 times the volume of FACS buffer 500 g, 3 min, wash the cells.
3.2.8 Resuspend cells to 106/1 ml with FACS buffer.
3.2.9 Perform flow cytometric sorting to set the gate using the non-transfected sample as a control. Positive cells are collected in separate tubes or multiwell plates containing a final concentration of 100 U/ml penicillin and 100 μg/ml streptomycin and sorted on the machine.
4 Further processing of sorted cells4.1 Functional analysis of interacting proteins
Since both bait and prey chimeric proteins were stably expressed in the screened clones, these clones were transfected with rPAP luciferase reporter genes that could be used to confirm bait-prey interactions. Together with these reporter genes, different receptor-decoy constructs were screened for a number of false positives after transfection. For example, when screening with a receptor-decoy chimera, this is chimeric with the extracellular portion of EPOR, then variants such as the chimera containing the extracellular portion of LR can be used to weed out non-EPO-dependent interactions. By co-transfecting with constructs that lack decoys, decoy-dependent protein interactions can be detected.
4.2 Recognition of prey
Prey-specific sequences were amplified by conventional methods, either by PCR with genomic DNA or by one- or two-step RT-PCR. PCR was performed with primers specific for the flanking and retroviral 3'-end repeat sequences of gp130. when used with the forward gp130-specific primer: 5' GGCATGGAGGCTGCGACTG-3', and the primer specific to the 3' end repeat sequence of retrovirus: 5'-TCGTCGACCACTGTGCTGGC-3', and the annealing temperature was 70 ℃, the PCR amplification could be carried out effectively. During the PCR process, care should be taken to avoid false positive bands due to contamination. Therefore, the operation should be carried out in a laminar flow cabinet, and all consumables and reagents should be free of template contamination. After separation by agarose electrophoresis, the specific bands are recovered and sequenced with gp130 specific primers.
For more product details, please visit Aladdin Scientific website.