D-Luciferin In Vitro Protocol
D-Luciferin In Vitro Protocol
Introduction
Luciferin is a widely used bioluminescent reporter for in vitro imaging of luciferase expression. This water-soluble substrate for firefly luciferase (typically from Photinus pyralis) uses ATP and Mg²⁺ as co-factors and emits a characteristic yellow-green light in the presence of oxygen. Since the reaction is ATP-dependent, it can also serve as a life-death indicator by signaling the presence of metabolic activity.
Firefly luciferase is among the most sensitive and user-friendly reporter genes for studying gene expression. It is commonly used to monitor promoter activity in bacteria, cultured cells, and transgenic plants or animals. The ~61 kDa monomeric enzyme is active without requiring post-translational modifications. By catalyzing the oxidative decarboxylation of luciferin, luciferase generates a bioluminescent signal with one of the highest known reaction efficiencies. When assay conditions are optimized, the emitted light correlates directly with transcriptional activity, making this system ideal for characterizing gene regulation via promoter/enhancer elements.
This handbook provides a straightforward, affordable, and reliable protocol for detecting luciferase activity in your in vitro system.

Bioluminescent Reaction

Sensitivity of the bioluminescent reaction can be influenced by temperature, pH, and substrate concentration. We recommend the following conditions for optimal results:
- Use a buffer with pH 7.8.
- Prewarm all reagents to room temperature before use.
- Include an excess of ATP and Mg²⁺ in the assay buffer.
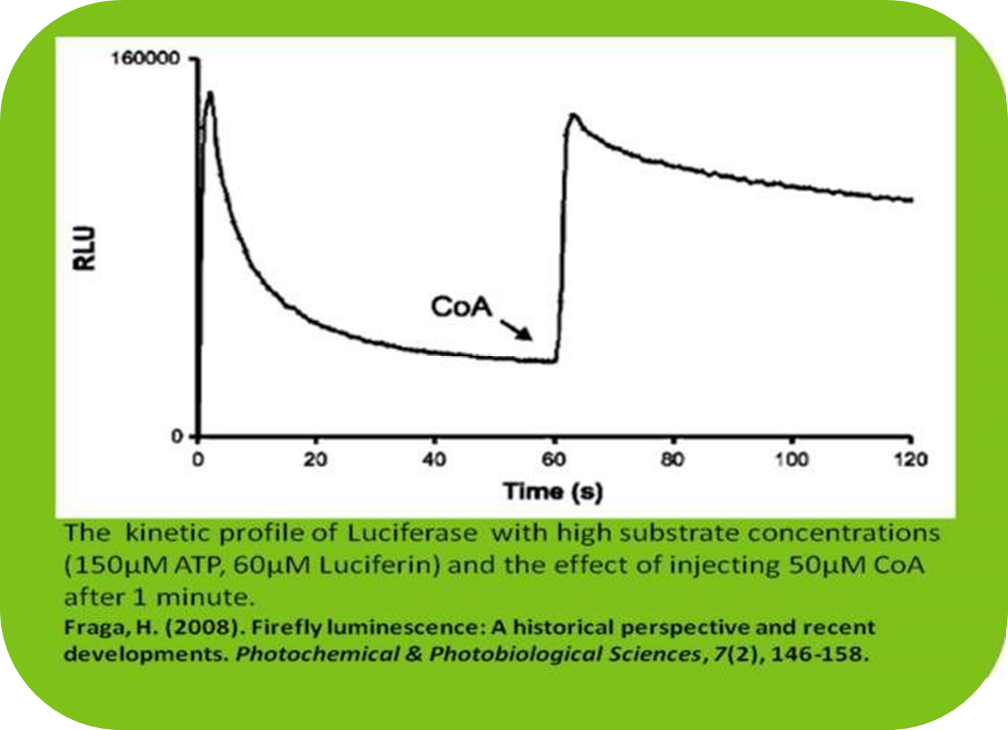
Upon addition of luciferin to a luciferase-containing sample, light emission peaks within 0.3–0.5 seconds and decays rapidly, with a half-life of approximately 0.5–1 minute. Coenzyme A (CoA), when included in the buffer beforehand, extends this half-life to 2–5 minutes. If added after the reaction has started, CoA can still promote secondary light flashes (Fraga, 2008).
Recommended Luciferase Assay Buffer (adapted from Oba et al., 2003):
- 100 mM Tris-HCl (pH 7.8)
- 5 mM MgCl₂-250 μM CoA
- 150 μM ATP (Fraga, 2008)
- 150 μg/mL D-Luciferin (Aladdin)
Optional additions include DTT or EDTA (Steghens, 1998), depending on your assay's requirements. A pilot test is encouraged to optimize the kinetic curve for your specific cell line.
Product Specifications
D-Luciferin, Potassium Salt
Aladdin Catalog # D1375449, D1375455, L120798
Molecular Formula: KC11H7N2O3S2
MW: 318.42 g/mol
D-Luciferin, Sodium Salt
Aladdin Catalog # D1375459, D1375464, D115509
Molecular Formula: NaC11H7N2O3S2
MW: 320.32 g/mol
Storage & Handling: Store at -20°C. Protect from light.
Materials
- D-Luciferin (Potassium/Sodium Salt)
- Luciferase Assay Buffer (TMCA)
- Tris-HCl (pH 7.8)
- MgCl₂
- Coenzyme A CoA (hydrate)
- ATP (disodium salt hydrate)
- Luciferin Stock Solution (LSS)
- PBS (without Ca²⁺ or Mg²⁺)
- Cell Lysis Buffer (Mammalian, Bacterial, Yeast)
- Luciferase (for calibration/control)
Luciferin Stock Solution (LSS) Preparations
1. Prepare 15 mg/mL (100x) LSS in molecular biology grade H₂O.
- Use immediately or aliquot and store at -80°C for up to 1 month.
2. Final concentration of Luciferin substrate, LSS , in the Luciferase Luminescence Assay:
- 150 μg/mL (471 μM for Luciferin-K, 468 μM for Luciferin-Na)
Luciferase Assay Buffer (TMCA) Preparation
Prepare individual components in molecular grade H₂O, then combine into TMCA. Adjust concentrations accordingly:
1. Prepare a 500mM MgCl2 (100x) solution.
a. Weigh 476.05 mg (95.21 g/mol) of MgCl2 and dissolve in 10 ml H2O.
b. Solution may be stored at RT.
2. Prepare a 25mM CoA (100x) solution.
a. Weigh 191.88 mg (767.53 g/mol) of CoA and dissolve in 10 ml H2O.
b. Prepare a 0.2 µm filter by drawing 5-10 ml of H2O through the filter.
c. Sterilize 100x CoA through pre-wet filter and store at -20°C.
3. Prepare a 15mM ATP (100x) solution.
a. Weigh 82.67 mg (551.14 g/mol) of ATP and dissolve in 10 ml H2O.
b. Store at -20°C.
4. Prepare 400mM Tris-HCl, pH 7.8 (4x) buffer.
a. Weigh 4.85 g (121.14 g/mol) of Tris Base and dissolve in 85 ml H2O.
b. Adjust pH with 1M HCl until pH 7.8 and bring volume to 100 ml with H2O.
5. Prepare fresh 2x TMCA working stock at room temperature.
a. Calculate the required TMCA for the experiment; combine all components to 2x working concentration (in H2O) (subsequently reduced to 1x in luciferase assay). Example (for 1 mL of 2x TMCA):
- 500 µL Tris-HCl (4x)
- 20 µL MgCl₂ (100x)
- 20 µL CoA (100x)
- 20 µL ATP (100x)
- Fill to 1 mL with H₂O
Optional: Add DTT or EDTA at this stage.
Cell Lysis Buffers
Here are some chocies of Lysis Buffers for your particular needs.
- Bacterial Cell Lysis Buffer
- Mammalian Cell Lysis Buffer
- Tissue Culture Lysis Buffer
- Yeast Lysis Buffer
Preparation of Cell Lysate for Luciferase Extraction
Adherent (Monolayer) vs. Nonadherent (Suspension) Cultures
Cell cultures can be broadly classified into two systems: monolayer cultures on a substrate (adherent) or suspension cultures (nonadherent). Most animal-derived cells are anchorage-dependent (except for hematopoietic or similar cell lines) and require a suitable substrate to support proper adhesion—commonly referred to as “tissue-culture treated” surfaces. However, many of these cell lines can also be adapted for suspension culture.
Suspension cultures do not require tissue-culture-treated vessels. However, the medium should be agitated to ensure adequate gas exchange and to prevent cell death. These cultures should be monitored daily using cell counts to assess growth and density. Dilution can be used to promote further proliferation.
Adherent cultures must be observed daily under an inverted microscope to assess confluence. Most should be subcultured once they reach 80–90% confluence to prevent nutrient depletion and subsequent cell death. Adherent cells require tissue-culture-treated dishes for optimal adherence and growth. While glassware may be used in place of disposable plasticware, all residual detergents must be completely removed, and proper sterilization performed before use.
Adhesion characteristics vary between cell lines. In most cases, trypsin (or another protease) is used to detach cells. For certain lines—especially when proteases may damage the cells or affect membrane markers/receptors—mechanical detachment using a cell scraper in a small volume of media may be preferred.
To Detach an Adherent Cell Culture
(Adapted from Methods in Molecular Biology, vol. 290: Basic Cell Culture Protocols)
1. Aspirate the culture medium once the cells reach the desired confluence. Rinse the monolayer with 2–3 ml of room temperature (RT) PBS (without Ca²⁺ or Mg²⁺) to remove residual medium.
2. Aspirate PBS and add 3–4 ml of RT trypsin–EDTA (T/E). Incubate at 37°C for 3–5 minutes. Monitor detachment under an inverted phase-contrast microscope.
3. Once cells begin detaching, transfer them to a centrifuge tube containing 6–7 ml culture medium (with serum to inhibit trypsin). To recover all cells, rinse the dish 1–2 times with 5 ml of the cell/medium mixture and add to the tube.
4. Count the cells using a hemacytometer, then proceed to the lysis step.
Note: The cells will be centrifuged and washed prior to lysis (see below).
Eukaryotic Cell Lysis (Mammalian or Yeast)
Preparation Before Use
Depending on the application, DTT and EDTA may be added to the lysis buffer to a final concentration of 5 mM. If divalent metal ions are necessary, omit EDTA and instead add an appropriate divalent salt
Protease Inhibition
If the inhibition of protease activity is required, add a cocktail of protease inhibitors to prevent protease activities during the extraction procedure (Protease Inhibitor Cocktail (Suitable for mammalian cell and tissue extract, EDTA Free, 100X), Aladdin #P775091, or Protease Inhibitor Cocktail (Suitable for fungal and yeast, EDTA Free, 100X), Aladdin # P775089).
Mammalian
1. Harvest cells by centrifuging at 200–500 × g for 5 minutes. Carefully discard the supernatant. For adherent cells, detach or scrape them (refer to section above), then collect by centrifugation and remove the supernatant.
2. Rinse the cell pellet with 5–10 ml of PBS (without Ca²⁺ or Mg²⁺). After removing the supernatant, gently tap the tube to dislodge the pellet. Resuspend the cells by pipetting gently up and down in 5–10 ml PBS. Centrifuge once again to collect the cells.
3. Remove the PBS wash completely and discard it.
4. Gently resuspend the pellet in any remaining volume of PBS using pipetting. Add Mammalian Cell Lysis Buffer to the cell pellet. For every 10 ml of dense suspension culture, use roughly 1 ml of Lysis Buffer.
Alternative approach: Add 1 ml of Lysis Buffer per 0.05 g of wet cell pellet. To obtain a more concentrated lysate, you may reduce the buffer volume accordingly. In such cases, a single freeze–thaw cycle can help achieve complete cell lysis.
5. Use a pipette to thoroughly mix the cells into a uniform suspension. Place the mixture on ice for 15–30 minutes, gently inverting the tube from time to time to facilitate lysis.
Note: A freeze–thaw step is not required, but performing one or two cycles is often beneficial and does not harm the lysate.
6. Centrifuge the lysate at 20,000 × g for 30 minutes using a refrigerated centrifuge. Collect the clear supernatant for downstream applications.
Note: The pellet may still contain nuclear and membrane-bound proteins, which can be extracted using specialized detergents if needed.
Yeast
1. Harvest yeast cells (culture OD₆₀₀: 1.5–2.0) by centrifuging at 5000 × g for 5–10 minutes. Resuspend the resulting pellet in an equal volume of Yeast Suspension Buffer. Add 1 µl β-mercaptoethanol per 100 µl of the yeast suspension.
2. Pipette gently up and down to achieve a uniform suspension. Incubate the mixture at 4°C for 5 minutes, then pipette once more to re-suspend the cells.
3. Gently flick the Zymolyase® vial to mix its contents. Add 10 µl of Zymolyase® for every 100 µl of yeast suspension. Mix the suspension carefully to distribute evenly.
4. Incubate at 37°C for 30 to 60 minutes. To monitor lysis progress, mix 25 µl of the suspension with 1 ml of Yeast Lysis Buffer and measure absorbance at 800 nm.
5. After incubation, centrifuge at 1,500 × g for 5 minutes. Carefully remove the supernatant, leaving the spheroplast pellet behind.
OPTIONAL: Add 5–10 volumes of Yeast Suspension Buffer to wash the pellet. Tap the tube gently to resuspend, then centrifuge again as before. Discard the supernatant.
6. For lysis, resuspend the spheroplast pellet in 2–3× its volume of Yeast Lysis Buffer. Pipette gently several times to mix. Periodically invert the tube and incubate on ice for 30 minutes.
Note: A brief incubation at 37°C (1–3 minutes) or mild sonication may further enhance lysis. Sonication is required if genomic DNA needs to be sheared.A higher buffer-to-pellet ratio improves lysis efficiency.
7. Centrifuge at 20,000 × g for 30 minutes at 4°C. Carefully collect the clear supernatant, which is now ready for analysis.
Note: Additional Yeast Lysis Buffer is available for purchase and can be used in downstream processes such as chromatography and dialysis.
Trademark Notice: Zymolyase® is a registered trademark of Kirin Brewery Co. Ltd.
Bacterial Cells
Preparation Before Use
Depending on the intended application, DTT and EDTA can be incorporated. Prepare the required volume of Bacterial Cell Lysis Buffer by adding DTT and EDTA to reach a final concentration of 5 mM each. If your application requires the presence of divalent metal ions, omit EDTA; instead, supplement with a suitable divalent salt to a final concentration of 5 mM.
Protease Inhibition – When preventing protease activity is necessary, add a protease inhibitor cocktail to protect proteins during the extraction process (refer to Protease Inhibitor Cocktail, suitable for bacterial cell extracts, EDTA-free, 100×, Aladdin #P776695).
Protein Extraction with Simultaneous Nucleic Acid Removal
1. Harvest Cells – Pellet bacterial cells (culture OD₆₀₀ between 1.5 and 3.0) by centrifuging at 5000 × g for 10 minutes. Resuspend the pellet in 5–10 times its volume of Bacterial Lysis Buffer (e.g., for a 25 µL pellet, use 125–250 µL buffer).
2. Initial Suspension – Gently pipette the sample up and down until it becomes uniform. Keep the suspension on ice or at 4 °C for 5 minutes, then pipette gently again to ensure complete mixing.
3. Add Lysozyme – Thaw and mix the lysozyme solution by vortexing. Add 5 µL lysozyme per 100 µL of cell suspension in Bacterial Lysis Buffer, then mix gently.
4. Incubation – Incubate the mixture at 37 °C for 30–60 minutes.
Optional: To monitor lysis, take 25 µL of the suspension, dilute in 1 mL of Bacterial Cell Lysis Buffer, and measure OD at 590 nm.
5. Complete Lysis – After incubation, invert the tube several times to finish lysis. If needed, further disrupt the cells by pipetting with a narrow-bore tip or passing through a 20-gauge syringe needle.
Note: Additional Bacterial Lysis Buffer can be purchased for downstream uses such as chromatography or dialysis.
6. Nucleic Acid Removal – During lysis, DNA and RNA are partially degraded, reducing lysate viscosity. Some DNA fragments may persist but generally will not hinder subsequent steps. For thorough nucleic acid removal, prepare the Bacterial Lysis Buffer without EDTA. After lysis, EDTA can be added to a final concentration of 2.5 mM.
7. Clarification – Centrifuge the lysate at 20,000 × g for 30 minutes at 4 °C. Collect the clear supernatant, which is now ready for analysis.
Protein extraction with spheroplast formation
Suitable when Lysozyme contamination must be avoided.
Follow bacterial protein extraction steps 1–4 from the method above, then proceed:
5. After the incubation period, centrifuge the suspension at 200–500 × g for 10 minutes. Gently remove and discard the supernatant, leaving the spheroplast pellet in the tube.
a. Optional – Re-suspend the spheroplast pellet in 5–10 volumes of Bacterial Suspension Buffer. Repeat centrifugation as above and discard the supernatant.
6. Lysis: Re-suspend the spheroplast pellet in an appropriate amount of Bacterial Lysis Buffer (approximately 2–3 times the pellet volume). Mix by pipetting up and down several times, inverting periodically. Keep the sample on ice for 30 minutes. To further enhance lysis, you may incubate the cells at 37 °C for 1–3 minutes or apply a brief sonication step. Note: A higher ratio of Bacterial Lysis Buffer to pellet volume will generally improve cell lysis efficiency.
7. Centrifuge the lysate at 20,000 × g for 30 minutes at 4 °C. Collect the clear supernatant (lysate), which is now ready for downstream analysis.
Isolation of Inclusion Bodies:
To isolate inclusion bodies, following the lysis step, centrifuge the bacterial lysate at 30,000 × g for 30 minutes at 4 °C. Carefully collect the resulting inclusion body pellet and wash it twice with Bacterial Lysis Buffer diluted 10-fold (for example, suspend the pellet in the buffer and centrifuge again to re-pellet the inclusion bodies). The cleaned inclusion bodies can then be used for solubilization and protein refolding.
Luciferase Calibration Curve
To accurately quantify luciferase levels in samples, it is essential to determine the linear detection range of your specific luminometer. This ensures reliable light intensity readings and avoids signal saturation at high luminescence levels.
1. Prepare a dilution series of luciferase—using either cell culture lysate or purified luciferase—in 1× lysis buffer (or PBS for purified luciferase) supplemented with 1 mg/ml BSA. (BSA helps stabilize the diluted luciferase enzyme.)
a. Include a control sample containing no luciferase to measure background luminescence.
Purified Luciferase Stock: Dissolve 1 mg luciferase in 100 mM Tris-HCl, pH 7.8, with 5 mM MgCl₂. The stock can be aliquoted and stored at –20 °C, or mixed with an equal volume of 100% glycerol and stored at –20 °C (glycerol stocks remain unfrozen at this temperature). Both frozen and glycerol-containing stocks remain active for at least two years under these storage conditions.
2. Dispense 100 µl of 2× TMCA (to achieve a 1× final concentration) into a cuvette or into wells of a 96-well plate.
3. Add 2 µl of 100× LSS (to achieve a 1× final concentration) to each sample.
4. Add 98 µl of the diluted luciferase stock or lysate to the cuvette or plate wells. Incubate at room temperature for 5–10 minutes, protected from light. (This step stabilizes luminescence, ensuring more accurate calibration curve measurements.)
a. For consistency, incubate all samples for exactly the same duration before reading.
b. Note: Volumes are calculated assuming the addition of 100× LSS to yield a final 1× TMCA concentration. Adjust the reaction mix proportionally for different total volumes.
5. Measure light output using a luminometer.
6. Generate the luciferase calibration curve and determine light output vs. cell number (or vs. µg luciferase).
Tip: If the assay does not produce a suitable linear relationship, lower the luciferin concentration.
Luciferase Luminescence Assay
The luciferase luminescence assay performance will be influenced by the type of luminometer used—whether it is a manual, single-tube with injector, or plate-reading luminometer. Always consult the user manual for your instrument regarding injection capabilities and programming
instructions.
Note: The concentration of LSS may need to be adjusted to meet the injection volume specifications of your particular luminometer.
In the assay mixture, the final concentration of TMCA should be 1×. Adjust the amounts of lysate and LSS so that the total volume corresponds to the intended final TMCA volume.
Example 1 – Manual Luminometer (final reaction volume: 200 µl)
1. Add 100 µl TMCA to a clean luminometer cuvette.
2. Add 50–98 µl lysate to the cuvette.
3. If necessary, add H₂O to bring the total volume to 198 µl.
4. Add 2 µl LSS (for a 1× final concentration) and read immediately on the luminometer.
Example 2 – Luminometer with Injection Capability (single sample or plate; final reaction volume: 200 µl)
1. Dispense 100 µl TMCA into a luminometer cuvette or each well of a 96-well plate (solid white, flat-bottom plates are recommended).
2. Add 50–98 µl lysate to the cuvette or well.
3. If required, add H₂O to reach 198 µl total volume.
4. Program the luminometer to inject LSS, apply a 2–5 second delay, and then record luminescence for 10 seconds.

References
Bai, X., et al. (2010). Both cultured and freshly isolated adipose tissue-derived stem cells enhance cardiac function after acute myocardial infarction. European Heart Journal, 31(4), 489-501.
Fraga, H. (2008). Firefly luminescence: A historical perspective and recent developments. Photochemical & Photobiological Sciences, 7(2), 146-158.
Helgason, C. D. and Miller, C. L. (2005). Methods in Molecular Biology, vol. 290: Basic Cell Culture Protocols, Third Edition. Humana Press.
Oba, Y., Ojika, M., & Inouye, S. (2003). Firefly luciferase is a bifunctional enzyme: ATPdependent monooxygenase and a long chain fatty acyl-CoA synthetase. FEBS letters, 540(1), 251-254.
Seliger, H. H., & McElroy, W. D. (1960). Spectral emission and quantum yield of firefly bioluminescence. Archives of biochemistry and biophysics, 88(1), 136-141.
Steghens, J. P., Min, K. L., & Bernengo, J. C. (1998). Firefly luciferase has two nucleotide binding sites: effect of nucleoside monophosphate and CoA on the light-emission spectra. Biochemical Journal, 336(Pt 1), 109.
Aladdin: https://www.aladdinsci.com/