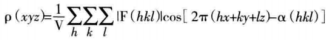posted this at Jun 9, 2025 Updated at Dec 23, 2024
Summary
The ability of protein molecules to crystallize has been reported for more than 170 years, and the discovery of X-rays by Rntgen WC in 1895 and X-ray diffraction from crystals by Laue MV in 1912 opened a new era in the study of crystal structures. In 1953, Perutz MF solved the phase problem of diffraction in the determination of the crystal structure of biological macromolecules by homocrystalline substitution of heavy atoms, and the low-resolution spatial structures of myoglobin and hemoglobin were obtained by Kendrew JC and Perutz MF in 1957 and 1959, respectively. Since then, X-ray diffraction has become an important tool for studying the spatial structure of biological macromolecules. The main steps of protein spatial structure analysis by X-ray crystallography include: (1) protein crystallization; (2) X-ray diffraction data collection; (3) calculation of electron density from diffraction data and phase information; and (4) establishment and modification of protein structure model. X-ray crystallography consists of five methods: protein crystallization, X-ray diffraction to analyze protein crystal structure, homocrystalline substitution, molecular substitution, and multi-wavelength anomalous scattering.
Principle
The basic principles of protein X-ray crystallography (X-ray crystallography) mainly include the preparation of protein crystals and the use of X-ray crystallography to analyze the spatial structure of protein crystals.
(I) Principle of protein crystallization
When the protein solution reaches the supersaturated state, the protein molecules in the random state will be changed into the ordered state and precipitated out from the solution, and this process of ordering is called protein crystallization. Due to the large molecular weight of proteins, complex geometry, diverse surface charges, local structure may have high flexibility, and some proteins are prone to aggregation and precipitation in solution, it is difficult to obtain molecularly ordered single crystals of proteins, which is still one of the bottlenecks in the analysis of crystal structure.
The crystallization process of protein can be divided into two stages, the first is the formation of nuclei, and then the surrounding molecules to the nucleus aggregation, the crystal gradually grows. The formation of nuclei of a certain size is the determining factor for crystal formation, and if the nuclei are too small they may dissolve. The key to protein crystallization technology is to control the degree of supersaturation of the protein solution and the speed of reaching supersaturation. If the protein solution is highly supersaturated, the protein molecules will precipitate as an amorphous precipitate. Protein crystallization should begin with a slow saturation of the protein solution, which leads to a suitable degree of supersaturation and the formation of a small number of nuclei. Protein molecules continue to combine to the nucleus, the degree of supersaturation of the solution is gradually reduced, and finally the solution and the crystal are in equilibrium.
(ii) Principle of protein crystal structure analysis by X-ray diffraction technique
The molecular arrangement of protein crystals is characterized by regularity, symmetry and periodicity. When X-rays enter the crystal from a specific direction, they interact with the electrons of the atoms in the crystal and scatter. The more electrons an atom has, the stronger the scattering ability. Electromagnetic waves scattered by the electrons of individual atoms in the crystal are coherently superimposed in space to form a diffracted beam. The direction of the diffraction line, i.e. the position of the spot on the diffractogram, is determined by the size and shape of the smallest repeating unit in the crystal, the cell; and the electrons of all atoms in the cell contribute to the intensity of the diffraction spot. Therefore, the direction of the diffraction line can be determined to determine the cell parameters, and the intensity of the diffracted spots can be determined to calculate the electron density distribution inside the cell by Fourier transform, and then the atomic space coordinates of the molecules inside the cell can be inferred from this.
The correspondence between the diffraction lines and the cell parameters can be given by either the Laue equation or the Bragg equation, and Bragg WL states that diffraction from a crystal can be seen as the reflection of an X-ray beam from a series of parallel planes in the crystal. Diffraction can only occur if the difference in the optical ranges of the reflected rays is equal to an integer multiple of the wavelength of the X-ray beam. Bragg's law gives the relationship between the X-ray wavelength λ, the angle of reflection θ, and the distance between the crystal planes, dhkl, as nλ = 2dhklsinθ. The X-ray wavelength, λ, is known, and the angle of reflection θ can be determined from the diffraction. The
reflection angle θ can be obtained from the diffraction data and hence the crystal plane spacing dhkl.
The value of the electron density p at the spatial coordinates (xyz) can be given by the following function:
where V refers to the total cell volume, IF(hkl)l is the structure amplitude, α(hkl) is the phase, and hkl is the miller indices. The electron density function is related to the structure amplitude and phase. In diffraction experiments, the intensity information of diffracted points can be recorded. The structure amplitude can be derived from the diffracted intensity. However, the phase of diffraction data cannot be obtained directly from experimentally collected data. One of the central problems in crystal structure analysis is to solve the phase problem. A series of methods have been developed to obtain the phase information.
1. Homocrystalline replacement method Two crystals are called homocrystalline if they have the same spatial symmetry and the same cell parameters. Without changing the molecular and crystal structure, protein crystals (parent) can be chemically modified (e.g., locally replaced by heavy atoms) to obtain derivative crystals. Since the introduction of heavy atoms only affects the diffraction intensity of the crystals, but does not change the diffraction direction, by comparing the diffraction images of the two (e.g., Patterson's function method), we can get the position information of the heavy atoms in the derivative crystals and the phase information of the protein parent.
2. Molecular replacement method For proteins or mutant proteins with high structural homology and chemical modifiers of proteins, the spatial structure of the proteins are often very similar, and only the structural details are very similar.
The spatial structures of proteins with high structural homology or mutant proteins as well as chemical modifications of proteins are often very similar to each other, only the structural details are different. In this case, the known homologous structure can be used as a model of the protein to be tested, and its structure factor can be calculated and compared with the experimental results to determine the phase. This method requires a high degree of structural similarity between the two.
3. Multi-wavelength anomalous scattering When the frequency of X-rays is close to the absorption band of the atomic spectrum, the X-rays are not only scattered, but also resonantly absorbed, which disturbs the normal scattering, and the scattering generated in this case is called anomalous scattering. The anomalous scattering effect is particularly pronounced for heavy atoms. For heavy atom derivatives similar to those described above, anomalous scattering of at least two wavelengths can also be used to obtain phase information.
For more product details, please visit Aladdin Scientificwebsite.
We use cookies to ensure the website functions properly and, where permitted, to improve your experience. You can manage your preferences at any time in Settings. Learn more in our Cookie Policy.
Shall we send you a message when we have discounts available?
Remind me later
Thank you! Please check your email inbox to confirm.
Products are supplied to verified businesses, institutions, and qualified professionals for research and development use only. Not for use in humans, animals, diagnosis, or therapy.