Human diseases number in the tens of thousands, yet only a fraction have effective treatments—and even fewer can be called “curable.” In many cases, current therapies primarily relieve symptoms or slow progression rather than addressing root causes. In recent years, as CRISPR gene-editing technology has entered the clinic, a new class of genetic medicines has begun to materialize. The first CRISPR therapies for sickle cell disease and transfusion-dependent β-thalassemia have been approved, offering near “root-cause” treatment concepts for these severe hematologic disorders and marking the first true entry of programmable gene editing into routine medical practice.
I. CRISPR: A Precise “Genetic Scalpel” Originating from Bacterial Immunity
CRISPR—short for “clustered regularly interspaced short palindromic repeats”—was first discovered in bacterial genomes and is considered a kind of “molecular immune system.” Bacteria use CRISPR to recognize and resist phage invasion. Its core components can be summarized as two parts:
1) A guide RNA that provides “navigation” and recognizes specific DNA sequences via base pairing;
2) A CRISPR-associated nuclease (e.g., Cas9, Cas12) that cuts DNA once directed to the target site.
By changing the sequence of the guide RNA, the same protein “tool” can be retargeted to different genes—one of CRISPR’s major advantages over earlier gene-editing tools. At the same time, optimized systems often achieve high sequence specificity, reducing off-target cutting risk. This combination of “programmability” and “relative precision” makes CRISPR a strong candidate platform for drug development.
II. First Clinical Indications: Sickle Cell Disease and Transfusion-Dependent β-Thalassemia
These two diseases share a core problem: severely impaired oxygen-carrying capacity of red blood cells, both rooted in hemoglobin.
1.Sickle cell disease (SCD)
Normal hemoglobin is a tetramer composed of two α and two β chains, uniformly distributed within red cells. In SCD, a specific mutation on the β chain promotes further assembly of tetramers into long, rod-like aggregates. These rods distort cell morphology into a sickle shape, making cells more fragile and prone to occluding microvessels, which triggers recurrent vaso-occlusive crises, ischemia, and chronic organ damage.
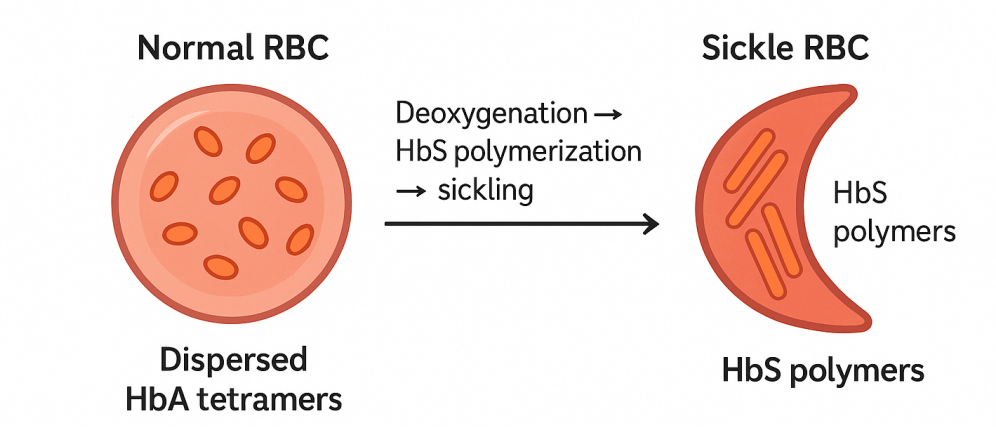
Figure1. Schematic of hemoglobin polymerization and red-cell sickling in SCD
2.Transfusion-dependent β-thalassemia (TDT)
Here, β-chain synthesis is severely reduced or absent, leading to markedly decreased total hemoglobin. Patients cannot sustain adequate oxygen transport and must rely on frequent transfusions.
Although one is a “structural abnormality” and the other a “deficiency in amount,” both share a physiological entry point: fetal hemoglobin. During fetal life, the body primarily expresses fetal hemoglobin; after birth, this is gradually replaced by adult α/β hemoglobin. Current CRISPR therapies are designed to: disrupt a key regulatory element (e.g., of BCL11A) in hematopoietic stem cells; lift repression of fetal hemoglobin so it re-expresses in adulthood; allow fetal hemoglobin to partially replace or compete with mutant β chains in red cells, thereby reducing rod-like aggregation and sickling, and increasing overall oxygen-carrying capacity in β-thalassemia. Clinical trials show that most SCD patients receiving such therapies experience long-term elimination of severe vaso-occlusive crises; most TDT patients can discontinue regular transfusions or see dramatically reduced transfusion needs. The magnitude of benefit far exceeds traditional symptomatic care.
III. Ex Vivo Gene Editing and Treatment Workflow
Currently approved CRISPR therapies use ex vivo editing of hematopoietic stem cells, with steps roughly as follows:
1.Collect hematopoietic stem cells: Enrich from bone marrow or peripheral blood and culture briefly ex vivo.After collection, viability assessment (e.g., Trypan Blue staining) is performed, followed by proceeding to the subsequent purification and editing workflows.
2.Ex vivo gene editing: Introduce the CRISPR system into these cells to target specific regulators so fetal hemoglobin re-expresses. Common electroporation/assembly buffers may contain HEPES, MgCl₂, and appropriate amounts of salt ions to maintain nuclease activity and cellular osmotic pressure.Edited cells can be screened and quality-controlled to confirm on-target editing and check for obvious off-target changes.
3.Conditioning and reinfusion: The patient receives high-dose chemotherapy to clear endogenous stem cells and “make space.” The edited stem cells are then reinfused.DMSO is commonly used for cryoprotection during short-term freezing/transportation and must be thoroughly removed prior to reinfusion.
4.Long-term hematopoietic reconstitution: The edited cells engraft and proliferate in the marrow, continuously producing red cells that express fetal hemoglobin. In principle, this is a one-time intervention with durable effect.
IV. Beyond Anemia: Expanding Disease Areas for CRISPR Therapies
The first therapies are only a starting point; ongoing clinical research is extending CRISPR to multiple disease domains.
1.Oncology: Enhancing immunotherapy
Multiple trials in leukemias, lymphomas, and solid tumors combine CRISPR with CAR-T cells—for example, editing T cells to improve tumor recognition; knocking out receptors that limit persistence or cause “exhaustion”; and building “off-the-shelf” CAR-T cells to reduce the cost and time of individualized manufacturing.
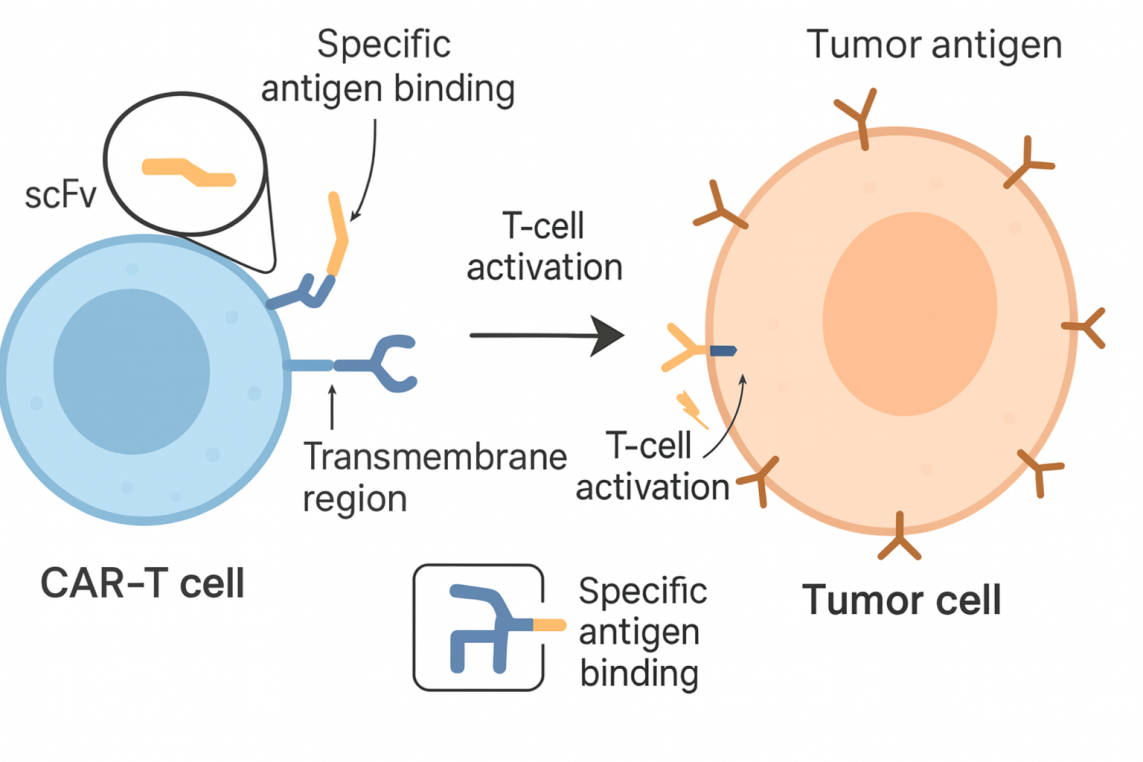
Figure 2. CAR-T cells recognizing tumor cells via chimeric antigen receptors
2.Inherited inflammatory diseases
In disorders such as hereditary angioedema, CRISPR-Cas9 is used to lower specific inflammatory mediators. Early results show some patients have almost complete cessation of attacks with good observed safety. Unlike hematologic diseases, these approaches have adopted systemic administration and in vivo editing—an important proving ground for feasibility.
3.Chronic bacterial infections
For antibiotic-refractory chronic urinary tract infections, researchers are using phages carrying CRISPR-Cas3 to target pathogens. Cas3 performs processive DNA degradation, effectively “shredding” bacterial genomes and killing the bacteria. Delivered intravesically, this may limit systemic effects. Efficacy is under further evaluation.
The approvals for CRISPR therapies in SCD and TDT symbolize programmable gene editing’s move from lab to routine care. They provide unprecedented hope for specific patient groups and lay technological and regulatory foundations for subsequent genetic medicines. Looking ahead, with safer nucleases, finer delivery systems, and growing clinical experience, CRISPR may expand from a few hematologic diseases to broader genetic, oncologic, infectious, and immune disorders. In parallel, how we balance innovation, risk, cost, and equity will determine how this technology ultimately integrates into everyday medical practice.
References
【1】Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The Protein Data Bank. Nucleic acids research, 28(1), 235–242.
【2】Berman, H., Henrick, K., & Nakamura, H. (2003). Announcing the worldwide Protein Data Bank. Nature structural biology, 10(12), 980.
【3】Bolton, W., & Perutz, M. F. (1970). Three dimensional fourier synthesis of horse deoxyhaemoglobin at 2.8 Angstrom units resolution. Nature, 228(5271), 551–552.
【4】Carroll D. (2017). Genome Editing: Past, Present, and Future. The Yale journal of biology and medicine, 90(4), 653–659.
【5】Cohen, J. The latest round in the CRISPR patent battle has an apparent victor, but the fight continues. September 11, 2020. Accessed July 17, 2024.
【6】Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., & Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science (New York, N.Y.), 339(6121), 819–823.
Aladdin: https://www.aladdinsci.com/