T cells recognize specific antigenic molecules (epitopes) through T cell antigen recognition receptors, and although the body's T cell pool can respond to a variety of antigenic epitopes, only T cells specific for a particular antigenic epitope are able to produce a response. Upon stimulation of T cells by binding to the MHC-antigen peptide complex on antigen-presenting cells, T cells can undergo a proliferative response and produce cytokines. Tetramer binding and enzyme-linked immunospot (ELISPOT) techniques have been used to detect the proportion of T cells in the T cell pool that bind to or secrete cytokines in response to stimulation with specific antigenic peptides.
Principle
The principle of using cellular tracer dyes to detect the frequency of proliferation of antigen-specific T cell precursors is to use flow cytometry combined with tetramer and cytokine staining techniques to detect the proliferative response of different T cell fractions in response to antigenic stimulation using tracer dyes, which is used to detect the frequency of T cell precursors in the T cell pools that are able to bind tetramers, secrete cytokines, and proliferate in response to a specific antigenic stimulus.
Operation method
Frequency of antigen-specific T-cell precursor proliferation detected using cellular tracer dyes
Principle
The principle of using cellular tracer dyes to detect the frequency of proliferation of antigen-specific T cell precursors is to use flow cytometry combined with tetramer and cytokine staining techniques to detect the proliferative response of different T cell fractions in response to antigenic stimulation using tracer dyes, which is used to detect the frequency of T cell precursors in the T cell pools that are able to bind tetramers, secrete cytokines, and proliferate in response to a specific antigenic stimulus.
Materials and Instruments
Equipment: Move The basic process of using cellular tracer dyes to detect the frequency of proliferation of antigen-specific T cell precursors can be divided into the following steps: Monocyte-containing lymphocytes (PBMC) are prepared from human peripheral blood using Ficoll-Hypaque density gradient centrifugation. Cell preparation was performed under aseptic conditions, and the final PBMC cell concentration in culture was 3 × 106/well, with a recommended starting cell number of (5-6) × 106 due to cell loss during labeling with tracer dyes and washing. When PBMC were isolated, they were stained with cellular tracer dyes (PKH or CFSE) prior to culturing, and the cell number was reduced to (5-6) × 106 due to the fact that the PKH26 (absorption spectrum PKH26 (absorption spectrum 551 nm/emission spectrum 567 nm) or PKH67 (absorption spectrum 490 nm/emission spectrum 504 nm) were chosen as tracer dyes due to their better compatibility with the spectra of other fluorescent dyes. At the end of the culture, cells can be stained for phenotypes, tetramers and intracellular cytokines. For intracellular cytokine staining, the cells must be restimulated with antigen for more than 16 h. The cells can be stained for intracellular cytokines by using a fluorescent dye. 1.1 PKH Staining Procedure 1.1.1 Wash the cells twice with serum-free medium and count them with a blood cell counter. Aspirate the supernatant during the second wash, retain 25-50 μl of serum-free medium and cell sediment, and gently resuspend the cells. 1.1.2 Dilute the PKH stock solution to twice the final concentration (8 μM if the final concentration is 4 μM) in another clean conical-bottomed tube with Diluent C from Sigma's Cell Census Plus kit. Diluted dye should be used immediately; unused dye will be discarded. The concentration of the dye should be pre-tested beforehand, both to ensure sufficient fluorescence intensity and cell viability after staining, and to ensure that the high intensity of fluorescence will not cause difficulties in spectral compensation with other fluoresceins, for human lymphocytes, the final concentration of the dye is about 4 μM. 1.1.3 Adjust the cell concentration to 2 × 107 cells/ml with Diluent C. The volume of the conical-bottomed centrifuge tube used should be more than six times the volume of the cell suspension; this diluent is designed for uniform distribution of this lipophilic dye, but it is damaging to the cells, and therefore the cells should be kept in this diluent for as short a time as possible. 1.1.4 Add an equal volume of 2 × PKH dye to the cell suspension and incubate at room temperature for 3 min. Since the labeling of the dye to the cells is very rapid (typically 80% within 15 s), it is important that the cells and dye mix gently and bind quickly to ensure uniform labeling of the dye. 1.1.5 After 3 min, an equal volume of human AB serum or fetal bovine serum (FBS) is added to the cell suspension to terminate the binding of the dye. 1.1.6 Wash 3 times with medium, count, and readjust the cell concentration to 1.5 × 106 cells/ml . 1.2 CFSE Staining Procedure 1.2.1 Dilute a 10 mM CFSE storage solution to 1 mM with PBS prior to cell labeling. 1.2.2 Wash the cells twice with serum-free medium and count them with a blood cell counter and adjust the cell concentration to 5 × 107 cells/ml using a conical-bottomed centrifuge tube more than 6 times the volume of the cell suspension. 1.2.3 Add 2 μl of CFSE (final concentration of CFSE is 2 μM) per ml of cell suspension. The concentration of the dye must be pre-tested and adjusted so that it has sufficient fluorescence intensity to maintain the viability of the stained cells (if the fluorescence intensity of the dye is too high, it will cause difficulties in compensating for the spectra of other fluoresceins). 1.2.4 Co-incubate the cells with the dye at 37 °C for 15 min with occasional mixing. 1.2.5 After 15 min, terminate the staining with 5 times the volume of pre-cooled medium containing 10% FBS. 1.2.6 Allow to stand at room temperature for 5~10 min. 1.2.7 Wash 3 times with medium and readjust the cell concentration to 1.5 × 106 cells/ml. 1.3 Cell Culture 1.3.1 After labeling the cells with a tracer dye, count the cells and check the cell viability using the Tepan blue reject method. 1.3.2 Add cells to a 24-well cell culture plate (3 × 106 cells/well) or a 12-well cell culture plate (5 × 106 cells/well) at a final cell concentration of 1.5 × 106 cells/ml. 1.3.3 Add antigenic peptides or proteins at the start of the culture at a concentration of antigen that ensures that it promotes cell proliferation; this concentration should be titrated beforehand by 3H doping assays or other dye dilution assays. 1.3.4 As a positive control for cell proliferation, add a final concentration of 5 μg/ml ConA or 2 μg/ml PHA to cultured cells that have not been stimulated by the addition of antigen, and incubate for 4-5 d. The final concentration of ConA should be 5 μg/ml ConA or 2 μg/ml PHA. 1.3.5 As a negative control for cell proliferation, cell culture wells do not contain mitogen or antigen. 1.3.6 Incubate the cell culture plate at 37 ℃, 5% CO2 for 6~10 d. The length of time depends on the intensity of the proliferative response, check the cultured cells regularly and add fresh medium when needed. 1.3.7 If phenotype, tetramer binding and intracellular cytokine staining are not performed, centrifuge the cells at the end of the culture and resuspend them with 0.2~0.4 ml of washing buffer and analyze them directly by flow cytometry. Correspondingly, 0.2~0.4 ml of 1% formaldehyde washing buffer can be added to the cells after washing, and then analyzed by flow cytometry after overnight fixation at 4 ℃.
① 12, 24, 96 well cell culture plates;
② 15 or 50 ml conical bottom centrifuge tube;
③ Centrifuge;
④ Colored fluorescent microspheres (RCP-30-5, Spherotech, Libertyville, IL);
⑤ Flow cytometer.
Reagents:
① Suitable lymphocyte culture medium (e.g., AIMV medium suitable for human cells (Gibco
① Suitable lymphocyte medium (e.g. AIMV medium for human cells (Gibco
Invitrogen Corporation, Carlsbad, CA) and serum);
② Antigenic proteins or peptides used for stimulation;
③ Knife bean protein A or phytohemagglutinin (PHA, Sigma, St. Louis, MO) used as a positive control stimulus for the proliferative response;
④ Tracer dyes: PKH26 or PKH67 [Cell Census Plus or Cell Linker kit (Sigma)], or diethylcarbamoyl succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR);
⑤ Fetal bovine serum (Sigma or others) or human AB serum (Nabi, Boca Raton, FL);
⑥ Brefeldin A (Sigma);
⑦ Fopperol myristate (PMA) and ionomycin (Ionomycin) (Sigma);
(viii) Wash buffer: PBS containing 1% bovine serum albumin and 0.1% sodium azide;
⑨ Human Globulin Cohn Components II and III (Sigma).
If intracellular cytokine staining is to be performed, antigenic stimulation must be added again on the last 1d of culture and treated with Brefel-din A to ensure that new cytokines are synthesized by the cells and stored in the cytoplasm for detection.
2.1 16 h before the end of cell culture, add antigen to stimulate the cells again (control does not need to be added).
2.2 At the same time, Brefeldin A at a final concentration of 1 μg/ml is added to the wells.
2.3 For the positive control wells of cytokine secretion, add PMA and 500 ng/ml Ionomy-cin at a final concentration of 10 ng/ml.
3. After the end of cell culture, the cells were optionally stained before flow cytometry analysisAt the end of cell culture, cells may be analyzed for tetramer binding and/or phenotypic staining, and if desired, cells may be stained for fixed, permeabilized, and intracellular cytokines.
1 Phenotypic analysis and/or tetramer binding staining
1.1 At the end of the culture, cells are collected and centrifuged.
1.2 Resuspend the cells with washing buffer (if intracellular cytokine staining is to be performed, add a final concentration of 1 μg/ml of Brefeldin
A) to re-suspend the cells, and the cell concentration was adjusted to (3~8) × 103 cells/0.03 ml.
1.3 Add an equal volume of human globulin (6 mg/ml for Cohn Component II and III stocks) to mix and seal non-specific Fc receptor binding.
1.4 Add cells [(3~8 ) x105 cells, 0.06 ml] to a 96-well plate, add 2~10 μl saturated concentration of tetramer, and incubate at 37 ℃ for 20 min.
1.5 Add 5~20 μl saturated concentration of phenotypic antibody, and act at 4 ℃ for 20 min.
1.6 If cytokine staining is not done, wash 3 times with pre-cooled washing buffer (200 μl/well), fix overnight at 4 ℃ with washing buffer containing 1% formaldehyde (0.2 ml/well), and perform flow cytometric analysis on the second day.
2 Intracellular cytokine staining
2.1 If intracellular cytokine staining is performed, wash with Brefeldin A wash buffer 3 times (200 μl/well), and then fix with 4% formaldehyde wash buffer for 20 min at room temperature.
2.2 After fixation, wash 3 times with washing buffer containing 0.1% saponin (for membrane penetration).
2.3 Resuspend the cells by adding 0.1 ml of washing buffer containing 0.1% saponin and add 10 μl of human globulin Cohn component (6 mg/ml).
2.4 Add 5-20 μl of saturated concentration of anti-cytokine fluorescein-labeled antibody or corresponding isotype control to each well, and incubate the cell culture plate at 4 ℃ for 60 min.
2.5 Wash twice with washing buffer containing 0.1% saponin, and then wash once with simple washing buffer.
2.6 Resuspend the cells with 0.2-0.4 ml of washing buffer containing 1% formaldehyde, incubate at 4 ℃ overnight, and then analyze the cells by flow cytometry on the second day.
4. Flow Cytometry4.1 The laser excitation of the flow cytometer and the band-pass filter of the photomultiplier tube (PMT) need to be matched with the selected fluorescent dyes. The excitation and emission spectra of PKH67 and CFSE have characteristics similar to those of green fluorescein, while PKF26 is similar to phycoerythrin (PE), and all three dyes can be excited by a 488 nm laser.
4.2 Fluorescence parameters including tracer dyes need to be logarithmically amplified. The PMT voltage parameter for detecting tetramer binding and cytokine staining for phenotyping needs to be set so that the negative cells are below the first of the 4 decimal scales, and the PMT voltage parameter for detecting tracer dyes needs to be set so that the nonproliferating cells are in the highest decimal scale (the appropriate position is about 10,000, when the scale of the relative fluorescence intensity is in the range of 1~10,000, and the relative fluorescence intensity is about 1~10,000, and the relative fluorescence intensity is about 1~10,000. 10,000, the appropriate position is approximately 5,000).
4.3 Forward and side scattering data are usually collected at linear amplification, and the voltage/amplification of the scattering parameter should be set so as to facilitate gating, i.e., to exclude dead cells and debris with low forward scattering light, but to include proliferating cells with high forward and side scattering light.
4.4 Spectral compensation adjustment is particularly important when performing multiparametric analysis, but will be difficult when PKH and CFSF fluorescence intensities are too bright, so the use of low concentrations of tracer dyes will facilitate compensation adjustment.
4.5 For accurate data analysis, it is important that enough cell data are captured in the data file so that the destination cell with the lowest frequency count has enough cells in it. For example, tetramer-positive cells or cells with darker PKH/CFSE or cytokine-positive cells are circled via a set gate, and a sufficient number of cells are collected into the data file so that there are no less than 500 cells in said gate. All data collected from cells are stored in a list mode file.
4.6 Data from colored fluorescent spheres with different fluorescence intensities must be collected into the data file at the same voltage/amplification parameter settings (changes in forward and side scattering affect the fluorescent spheres above the threshold for side scattering).
5. Data analysisPrior to data analysis, cells were gated based on cell phenotype and/or cytokines and tetramers. For proliferation and precursor frequency analysis, ModFit software for proliferation analysis is available for PKH and CFSE dye analysis. ModFit 3.1 or higher will be able to calculate precursor frequencies.
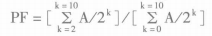
For more product details, please visit Aladdin Scientific website.