PCR-single-stranded conformational polymorphisms
PCR-single-stranded conformational polymorphisms
PCR-single-strand conformation polymorphism is a technique based on this DNA single-strand conformation property combined with gel electrophoresis to detect genetic variation. Currently, there are 1 main method applied to PCR-single-strand conformation polymorphism: PCR-single-strand conformation polymorphism.
Principle
The basic principle of PCR-single-strand conformation polymorphism is that under nondenaturing conditions, a single strand of DNA can fold itself to form a conformation with a certain spatial structure; this conformation is determined by the order of the bases in the single strand of DNA, and its stability is maintained by the interaction forces within the molecule (mainly hydrogen bonds).
Single-stranded DNA of the same length has a different conformation depending on the order of its constituent bases or even on the individual bases. Since polyacrylamide gel electrophoresis has extremely high resolution and resolution, the mobility of DNA single strands in neutral polyacrylamide gel electrophoresis without denaturant is more dependent on the conformation formed by the DNA single strands, in addition to the length of the DNA.
After the PCR product of DNA or RNA is denatured, when the target DNA or RNA is changed due to single base substitution, base insertion or base deletion, its single-stranded conformation also changes, and this conformational change can be reflected by the change of mobility in neutral polyacrylamide gel electrophoresis.
Schematic diagram of the principle of PCR-single-strand conformation polymorphism:
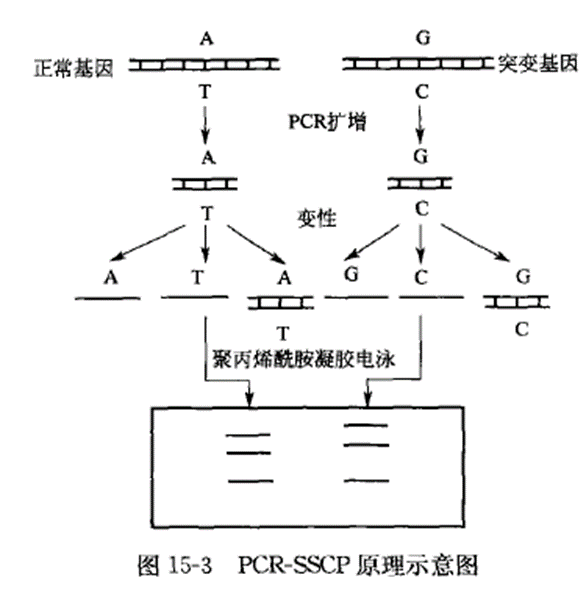
Appliance
The common application fields of PCR-single strand conformation polymorphism are as follows: 1. Application of PCR-SSCP in disease diagnosis Since the establishment of SSCP technology by Orita et al. in Japan in 1989, SSCP technology has received more and more attention as a means of detecting gene mutations.Combining with PCR technology, SSCP technology analyzes amplified DNA sequences by SSCP, which can be used to Combined with PCR technology, SSCP analysis of amplified DNA sequences can rapidly detect known mutations and screen for unknown mutations, and is widely used in cancer research and disease diagnosis. The use of silver staining instead of isotope labeling makes PCR-SSCP more rapid and convenient for clinical diagnosis. Zhu Bin et al. applied this method to detect Leber's hereditary optic neuropathy; the gene is located on chromosome 11, and the patient's gene has a GA mutation at locus 778; primers were designed to amplify a 340-bp fragment between 641 and 980; the results of SSCP analysis and silver staining showed that the bands at the front of the gel were undenatured double-stranded DNA, and the normal single-stranded DNA was slower than the mutated DNA. The swimming speed of normal single-stranded DNA is slower than that of mutated DNA, and two normal single-stranded and two mutated single-stranded genes can be detected by electrophoresis of heterozygous genes. This shows that SSCP technology can rapidly detect diseases and determine whether the mutant gene is in a pure or heterozygous form. Since the application of PCR technology can reduce the amount of samples, it can be used in early embryonic detection, thus preventing the birth of babies with certain diseases and playing an important role in eugenics. Lin Jinrong et al. established a non-invasive screening method for colorectal cancer by PCR-SSCP. Early diagnosis and timely treatment of the disease was achieved by detecting mutations in the APC (abenomatous polyposis coli) and MCC (mutated in colorectal cancer) genes in feces, which plays a key role in increasing the cure rate, prolonging the life of patients, and improving the quality of patients' survival.2 . Application of PCR-SSCP in taxonomic research PCR-SSCP is a more refined classification technique, which can distinguish the difference of individual nucleic acids of a gene, and has been widely applied to the classification of bacteria, viruses and parasites, etc. Vazquez used PCR-SSCP to analyze the four components of the Tep5-β gene family of Verticillium graminearum, and successfully identified the compound gene family. Vazquez used PCR-SSCP to analyze four components of the Tep5-β gene family of Verticillium graminearum, and successfully identified different forms of this complex gene family and polymorphisms in different strains of the parasite in the natural state, which opens up a new way for the classification of parasites.Koenig et al. analyzed the specific fragments of different genomic fractions of the beet necrotic yellows vein virus (SNV) from different countries after amplification by SSCP analysis, and according to the differences of the SSCP profiles, different isolates were classified into the three clusters of A, B, and P; of which The isolates with fewer SSCP bands found only in Pithirers, France, were localized to the P strain group, those found only in France and some parts of Germany to the B strain group, and the rest to the A strain group.
Operation method
PCR-single-stranded conformational polymorphisms
Principle
The basic principle of PCR-single-strand conformation polymorphism is that under nondenaturing conditions, a single strand of DNA can fold in on itself to form a conformation with a certain spatial structure. This conformation is determined by the DNA single-stranded bases, and its stability is maintained by local order interactions (mainly hydrogen bonds) within the molecule. The same length of DNA single chain with different order, or even a single base is different, the conformation formed by different, electrophoretic mobility is also different. PCR product denaturation, single-stranded product by neutral polyacrylamide gel electrophoresis, the target DNA containing a single base substitution, or several base insertion or deletion and other changes, due to changes in mobility will be the swim shift, so as to differentiate the mutant DNA from the normal DNA. Thus, PCR-SSCP analysis is a DNA single-stranded gel electrophoresis technique, which detects genetic variation based on the electrophoretic mobility changes of equal-length DNA single strands forming different conformations in a neutral polyacrylamide gel.
Materials and Instruments
Equipments: PCR instrument, electrophoresis instrument, radiation autoradiograph Move The basic process of PCR-single-strand conformation polymorphism can be divided into the following steps: The reaction was terminated at 37 ℃ for 30 min, 75 ℃ for 2 min and stored at 4 ℃. Put the gel into the color development plate, add 100 ml of 30% Na2CO3 (containing 10 μl of 37% formaldehyde), 50 ℃ warm bath color development, gently shake the color development plate, so that the silver particles on the sample band on the gel to restore uniformity, within a few minutes the staining band can be seen, and continue to warm bath until the electrophoresis band is clear. Add 5 ml of 2.3 mol/L citric acid buffer to terminate, rinse with distilled water several times, and take pictures.
Reagents:
1. PCR reaction reagents
Taq DNA polymerase, T4 polyribonucleotide kinase, buffer, [γ-32P] ATP and [α-32P] dCTP, primers and dNTP.
2. Electrophoresis reagents
① Acrylamide methacrylamide (49:1), deionized formamide, glycerol.
② Sampling buffer: 95 % formamide, 20 mmol/L EDTA pH8.0, 0.05% xylene cyanide, 0.05% bromophenol blue.
③ TAE buffer or TBE buffer.
3. Radiographic autoradiography and staining reagents
Radiographic autoradiography X-ray film.
② EB staining: 0.5 μg/ml EB.
③Silver staining reagents: methanol, acetic acid, glutaraldehyde, AgNO
3
Na
2
CO
3
, formaldehyde, citric acid.
(i) PCR amplification of genes
(1) PCR reaction without isotope labeling PCR reaction system: 10 × PCR buffer 5 μl, dNTP mixture 4 μl (2.5 mmol/L), primer 11 μl (final concentration 0.2 ~ 1.0 μmol / L), primer 21 μl (final concentration 0.2 ~ 1.0 μmol / L), Taq enzyme 0.1 ul (5 U/μl), template DNA 1 μl (<1 pg), and ddH2O was added to 50 μl.
Cycling reaction parameters were determined according to the characteristics of the primers and the genes to be amplified, and the reaction was cycled 30 times.
The amplification products were purified by PCR recovery kit and stored at 4 ℃.
(2) [γ-32P] ATP isotope-labeled PCR reaction
① PCR primer labeling:
10× T4 polynucleotide kinase buffer 2 μl
Primer 1 (10 μmol/L) 1 μl
Primer 2 (10 μmol/L) 1 μl
[γ-32P] ATP (5000 Ci/mmol) 4 μl
T4 polynucleotide kinase (5 U/μl) 4 μl ddH2O to 20 ℃. l
ddH2O to 20 μl
② PCR reaction (with labeled primers)
Reaction system:
10×PCR buffer 2 μl
Primer 1 (4~20~1.0 μmoI/L) 1 μl
Primer 2 (4~20~1.0 μmoI/L) 1 μl
dNTPs (2.5 mmol/L) 4 μl
Taq enzyme (5 U/ μl) 0.1 μl
Template DNA (<1 μg/μl) 1 μl ddH2O to 20 μl. l
ddH2O to 20 μl
for PCR amplification.
The product was purified by the recovery kit and stored at 4 ℃.
(3) [α-32P] dCTP isotope-labeled PCR reaction
Reaction system:
10×PCR buffer 8 μl
Primer 1 (4~20~1.0 μmoI/L) 1 μl
Primer 2 (4~20~1.0 μmoI/L) 1 μl
dNTPs (2.5 mmol/L) 4 μl
[α-32P] dCTP (3000 Ci/ mmol) 2 μl Taq enzyme μl
Taq enzyme (5 U/μl) 0.2 μl
Template DNA (<1 μg/μl) 1 μl
ddH2O to 80 μl
PCR amplification was performed.
The products were purified by the recovery kit and stored at 4 ℃ for backup.
(ii) Denaturation of PCR samples
(1) Alkaline denaturation: take 12 μl of PCR product, add 0.5 mol/L NaOH 1 μl and 20 mmol/L EDTA 1 μl, mix well, denature at 42 ℃ for 5 min, and then add the sample buffer.
(2) Thermal SDS denaturation method: Take 1 μl of PCR product, add 1 μl of 0.2% SDS solution and 1 μl of 20 mmol/L EDTA, mix well, and put it into the ice bath immediately after 3~5 min of boiling water bath and then cool it down for 3~5 min, then centrifuge it quickly for a moment and put it on the ice.
(iii) Preparation of polyacrylamide gel
Prepare polyacrylamide gel for routine electrophoresis.
(D) Color development or imaging of the gel
(1) X-ray film development
After electrophoresis separate the two glass plates carefully so that the gel is attached to one of them. Cut a piece of filter paper equal in size to the gel and lay it flat on the surface of the gel and carefully remove the glass plate. Place the filter paper and gel together in the X-ray dark box with the filter paper facing down and the gel side facing up. Wrap the gel and filter paper in plastic wrap, then place the film on top of the plastic wrap and add a sensitizing screen to the film. The dark box was placed at -70 ℃ for 36~72 h of radiographic self-development, and the film was developed with developer and fixer.
(2) EB staining
After electrophoresis, the gel was gently removed from the glass plate and immersed in 0.5 μg/ml EB staining solution prepared from buffer, and then stained at room temperature for 30~45 min, and then gently rinsed with water. The staining was rinsed gently with water and then observed and photographed on a UV detector.
(3) Silver staining method for color development Two methods are briefly described below.
①Carefully remove the glass plate and make the gel adhere to a glass plate, soak it in 10% acetic acid and 50% formic acid for 30 min, and shake it slowly. The gel and glass plate were then transferred to 7% acetic acid and 50% methanol for 30 min, and finally to 10% glutaraldehyde for 30 min with gentle shaking to fully fix the sample. The excess fixative was washed away with distilled water. Soak the gel in 0.1% AgNO3 solution for 30 min, and shake gently to distribute the silver particles on the gel.
② Put the gel into 50% methanol and 12% acetic acid solution for 10 min, then 10% methanol and 5% acetic acid for 10 min, and then 0.0034 mol/L potassium dichromate and 0.0032 mol/L nitric acid solution for 5 min. Put the gel into 0.012 mol/L silver nitrate solution for 20 min, and then put it into 0.28 mol/L sodium carbonate and 0.5 ml sodium carbonate and 0.5 ml sodium carbonate. Sodium carbonate and 0.5 ml/L formaldehyde were used to develop the color for about 5 min, and the reaction was terminated with 1% acetic acid under light condition. Note that the gel should be rinsed with deionized water after each step.
(4) Analysis of results
After denaturation and electrophoresis of the PCR samples, there are not necessarily only two single-stranded bands, due to the diversity of single-stranded conformation and the existence of intermediate structures, there can be more than one single-stranded band, but the single-stranded bands of different DNA fragments of different samples are the same color shade. However, the color of the single-stranded bands is consistent from one sample to another. The dynamic shifts representing the genetic variation may not necessarily appear in the two single-stranded bands, and the position of the single-stranded bands and double-stranded bands should be established according to the control samples in the experiment, so as to detect the dynamic shifts of the mutated DNA.
False positives cannot be ruled out when PCR-SSCP is used to detect point mutations in DNA samples. Mutations that cannot be detected in this way can be achieved by changing the experimental conditions or detection method, and the final screening of the swimming variant will determine that a mutation has occurred in the DNA fragment.
Caveat
1. Length of PCR amplified fragmentsPCR-SSCP has high requirements for the length of PCR amplified fragments when used to detect point mutations. When the amplified fragment is less than 400 bp, single strand separation by gel electrophoresis is more effective. PCR-SSCP can detect more than 90% of single base substitutions when the amplified fragment is around 200 bp, and around 80% of single base substitutions when the amplified fragment is 400 bp. When the amplified fragments are long, PCR-SSCP can be performed after digestion with restriction endonuclease, which will give better results.
2. Processing of PCR samples and productsThe purity of the sample DNA for PCR amplification should be high, with OD260/OD280 >1.
6, if not, it should be purified, and the specificity of the PCR amplified bands should be high; we can select the specific primers through the analysis of primers or change the conditions of PCR annealing temperature and time to achieve the specificity of the amplification, and it is better to purify the PCR products before sampling, which can avoid the damage of primers, dNTP and other substances. PCR products should be purified before sampling, so as to avoid the influence of primers and dNTP, etc. PCR products must be reasonably diluted (1:4 or 1:6, etc.), to avoid the single-stranded bands being blurred due to the recombination of the high concentration of DNA.
3. Preparation before electrophoresisThe thickness of the PCR-SSCP gel should not be more than 0.4 mm, and the sample wells should be carefully rinsed before sampling. Before filling the gel, note that the gel must be mixed well, the gel plate must be washed, and the gel must be filled quickly. It is better not to prepare the mother liquor of electrophoresis buffer for more than one month, and replace the newly diluted buffer every time of electrophoresis. This can reduce the smile-shaped band or wave-shaped band.
4. Gel concentration and crosslinking degreeDifferent concentrations of gels have their own effective separation ranges. 5% acrylamide has an effective separation range of 80-500 bp, 8% acrylamide has an effective separation range of 60-400 bp, and 12% acrylamide has an effective separation range of 40-200 bp. Commonly used gels are generally in the range of 5% to 8%, and it is necessary to decide the concentration of the gel according to the actual situation of your own in order to achieve the ideal separation effect. The concentration of commonly used gels is usually 5%~8%.The degree of cross-linking can be expressed as the percentage of methylenebisacrylamide in the total acrylamide monomer concentration. Gels with low cross-linking are favorable for the separation of complementary strands of DNA. The higher the percentage, the harder the gel and the smaller the pore size; conversely, the lower the percentage, the softer the gel, the larger the pore size, and the more sensitive it is to DNA conformation.The cross-linking degree of the gel in SSCP analysis is usually 1% ~2%. The ratio of acrylamide to methylenebisacrylamide is 49 : 1.
5. Concentration of glycerolLow concentrations of glycerol (5% ~10%) increase the efficiency of isolating mutant sequences. Because glycerol is a weak denaturant for nucleic acids, it partially opens the folded structure of the DNA single strand, thereby increasing the exposed surface area of the DNA molecule, and thus increasing the chances that the gel will be able to localize the structural differences caused by the mutation.Usually, the addition of glycerol at 20~25 ℃ gives satisfactory electrophoretic results. However, the viscosity of glycerol is high, and even higher at 4 °C, thus reducing the mobility of the molecules. Due to the different sequences of target genes, electrophoresis at 4~10 ℃ without glycerol gives better results. However, for the isolation of some DNA fragments in the presence of glycerol-free gels, the shifts in motion caused by the mutated sequences are rare. It is important to note that a very small number of mutations can only be detected at 25 °C under specific conditions without glycerol.
6. TemperatureDuring gel electrophoresis, the temperature of the gel plate increases as the electrophoresis time increases, which can be detrimental to the efficient isolation of mutant DNA. Therefore, it is important to take measures to control the temperature of the gel plate during the experiment. Air or circulating water cooling can be used to maintain a constant temperature. Usually, the temperature of the gel is controlled at 4 ℃ or 20~26 ℃.
7. VoltageThe effect of voltage on gel electrophoresis may be created by changing the temperature. Better results can be obtained at low voltage for a long period of time. Some experimentalists have also used electrophoresis conditions in which the voltage is firstly high and then lowered, which also gives good results.
8. Gel electrophoresis methodGenerally SSCP analysis is applied to plate polyacrylamide gel electrophoresis, supplemented by radioactive autoradiography or silver nitrate staining and other techniques to show the results. At present, many domestic and foreign researchers use capillary polyacrylamide gel electrophoresis to study SSCP; when using capillary electrophoresis for PCR-SSCP, fluorescence detection method is usually used.The fluorescence detection method is highly sensitive and specific, and the results are easy to read. However, fluorescent labeling prolongs the experimental process and costs more. These factors are not conducive to the popularization of capillary electrophoresis, but in practice, it can also be detected directly by UV detector, which can also detect the double-stranded and single-stranded DNA peaks of PCR products. This method is simple and easy to promote.
For more product details, please visit Aladdin Scientific website.