PCR-based large fragment DNA synthesis assay
PCR-based large fragment DNA synthesis assay
Chemical synthesis of DNA sequences provides a powerful tool for high level expression and functional studies of genes in heterologous systems. In recent years, many methods have been developed for synthesizing and collecting DNA sequences.
Early methods were mainly enzyme ligation and FokI, later self-initiated PCR, PCR pooling and template-directed ligation techniques were developed, and more recently methods for synthesizing and collecting long DNA sequences have been reported, typical of which are thermodynamically balanced inside-out (TBIO), double asymmetric (DAB), thermodynamically balanced inside-out (TBIO), and double asymmetric (DAB) methods. Typical methods include thermodynamically balanced inside-out (TBIO) PCR, dual asymmetrical PCR (DA-PCR) and overlap-extension PCR (OE-PCR), a two-step gene synthesis method based on sequential PCR.
Currently, there is one main method for PCR synthesis of large DNA fragments: PCR-based accurate synthesis of long DNA sequences.
Principle
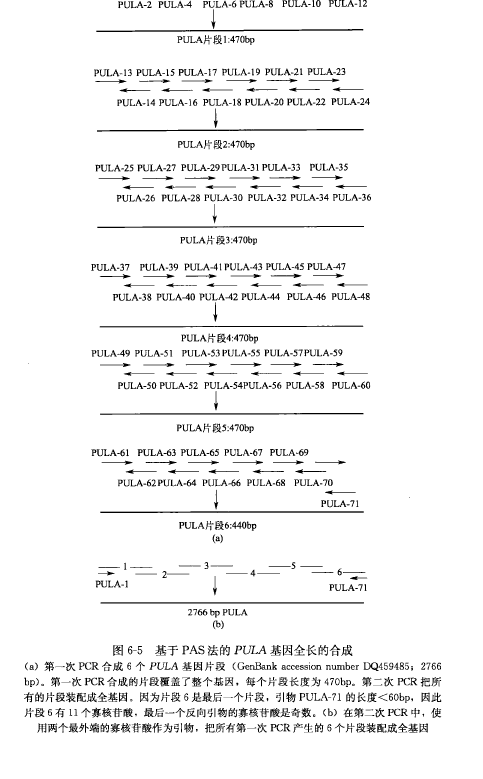
The basic principle of the PCR-based method for accurate synthesis of long DNA sequences is. Based on the target DNA sequence, design and chemically synthesize 60 bp long oligonucleotides with 21 bp overlap with neighboring nucleotides. To minimize errors in chemical synthesis, all oligonucleotides should be purified by PAGE. A set of 12 oligonucleotides of 60 bp adjacent to each other is synthesized by PCR into DNA fragments of 400-500 bp in length [Fig. 6-5 (a)]; most of the 5' forward oligonucleotides and the 3' reverse oligonucleotides (external oligonucleotides) are synthesized by using the 10 internal oligonucleotides retained as templates, using a high fidelity DNA polymerase (e.g., Pfu, less than 1 error per 1000 bp). errors per 1000 bp).
Since the concentration of the two external oligonucleotides is 20 times higher than that of the internal oligonucleotides, most of the PCR product should be about 400-500 bp DNA fragments. In the second PCR step, all 400-500 bp DNA fragments synthesized in the first PCR step are used as templates to synthesize a full-length DNA sequence [Figure 6-5(b)]. DNA polymerases that produce long, accurate DNA fragments should be used in this PCR step (e.g., sodium pyroborate DNA polymerase, which is capable of synthesizing fragments up to 10 kb in length and has a low error rate similar to that of Pfu). Finally, the synthesized gene sequences are identified by DNA sequence analysis, and errors in the sequences are corrected by overlap extension PCR.
Operation method
Accurate PCR-based synthesis of long DNA sequences
Principle
The basic principle of the PCR-based method for accurate synthesis of long DNA sequences is. Based on the target DNA sequence, design and chemically synthesize 60 bp long oligonucleotides with 21 bp overlap with neighboring nucleotides. To minimize errors in chemical synthesis, all oligonucleotides should be purified by PAGE. A set of 12 oligonucleotides of 60 bp adjacent to each other is synthesized by PCR into DNA fragments of 400-500 bp in length [Fig. 6-5 (a)]; most of the 5' forward and 3' reverse oligonucleotides (external oligonucleotides) are synthesized using the 10 internal oligonucleotides retained as templates, using a high fidelity DNA polymerase (e.g., Pfu, less than 1 error per 1,000 bp). errors). Since these 2 external oligonucleotides are 20 times more concentrated than the internal oligonucleotides, most PCR products should be approximately 400-500 bp DNA fragments. In the second PCR step, all the 400-500 bp DNA fragments synthesized in the first PCR step are used as templates to synthesize a full-length DNA sequence [Figure 6-5(b)]. In this PCR step, a DNA polymerase that produces long, accurate DNA fragments should be used (e.g., sodium pyroborate DNA polymerase, which is capable of synthesizing fragments up to 10 kb in length and has a low error rate similar to Pfu). Finally, the synthesized gene sequence is identified by DNA sequence analysis, and errors in the sequence are corrected by overlap extension PCR.
Materials and Instruments
Equipment: Move The basic process of PCR-based accurate synthesis of long DNA sequences can be divided into the following steps: 1. Synthesize a 400-500 bp DNA fragment from a set of 12 oligonucleotides of 60 bp next to each other (Figure 6-5). All reaction reagents should be stored and handled on ice throughout the process. For the PULA gene, which is 2,766 bp long, all synthesized oligonucleotides are divided into 6 groups for each DNA fragment [see Figure 6-5(a)]. 2. For each group, mix the 10 internal primers: 1 μL (30/μmol/L) of each primer is added to an EP tube and mixed gently. Positive control was "known template", in which 10 internal oligonucleotides were replaced with the appropriate sequence of the full-length gene; negative control was "no template", in which the internal oligonucleotides were replaced with water; at the same time, one primer from each pair of external primers, i.e., "single primer", was used as the negative control. Take PULA gene as an example, tubes A1, A2, A3, A4, A5 and A6 contain 10 internal oligonucleotides. 3. Remove 0.5 μL of the oligonucleotide mixture and add to a 500 μL PCR tube. For the PULA gene, remove 0.5 μL from tubes A1, A2, A3, A4, A5, and A6 to the corresponding tubes B1, B2, B3, B4, B5, and B6 [Figure 6-5(a)]. 4. Add 1 μL of each external primer (30 pmol) to each tube containing all 12 oligonucleotides for synthesizing a 400~500 bp DNA fragment. 5. Then add the following PCR reactants to each tube (containing a total of 2.5 μL of the 12 oligonucleotide mixture): Flick the contents through the tubes to mix and centrifuge. 6. PCR was performed under the following conditions (GeneAmp PCR System 9 600): 7. 10% (5 μL) of the PCR product was removed from each tube and subjected to agarose gel (1.0%) electrophoresis at 150 V for 20 min; the PCR product was quantified, and at least 100-200 ng of single-stranded DNA product was obtained from the first step of PCR to ensure that high-quality product was obtained from the second step of PCR. If multiple bands are seen in the first PCR step, the target gene should be gel purified. To synthesize long DNA sequences (e.g., 4 kb), synthesize a 2-kb intermediate before collecting the full-length DNA, clone and assay the intermediates, and correct them for errors before proceeding.
① EP tube;
② PCR (GeneAmp PCR System 9600);
③ ABI PRISM 310 bioanalyzer, etc.
Reagents:
① BigDye Terminator v3.1 Cycle Sequencing Method Kit;
② Oligonucleotide mixture.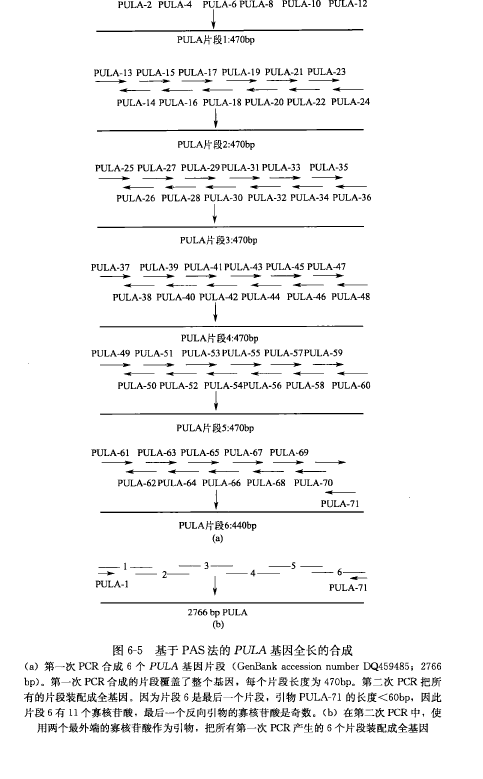


8. For all PCR products from step 7, mix one column of each product (50 ng of DNA) into a 500-μL EP tube (Figure 6-5). Note: Add approximately equal amounts of each product from the first PCR step to obtain high quality full-length DNA for the second PCR step.
9. Then remove 1 μL of the mixture and add it to a 500 μL PCR tube.
10. Add the two outermost primers (30 pmol) of the full-length DNA, e.g., PULA gene, oligonucleotide 1 and oligonucleotide 71 [Figure 5-6(b)]. Also add the following PCR reactants for PCR:

11. PCR (GeneAmp PCR System 9 600) was performed under the following conditions: "known template" as positive control, i.e., the full-length gene was used as template instead of the DNA fragment obtained in the first step of PCR; "no template", i.e., water was used instead of the template, and "single primer" was used as negative control.
12. All of the PCR products are subjected to agarose gel electrophoresis on a 1.0% agarose gel at 150 V for 20 min for gel purification. A clean, single band of the correct size should be obtained. The DNA fragment product from the second PCR step should be purified by gel separation to make later cloning easier. Note: The accuracy of the synthesized product should be verified and corrected for errors.
13. The PCR product is cloned into any vector and analyzed for DNA sequence. Note: Sodium pyroborate polymerase produces a flat-ended PCR product, and 3'-aminopurine must be added if a TA PCR cloning kit is used.
To ensure that the synthesized DNA is free of errors, sequencing is performed using the BigDye Terminator v3.1 Cycle Sequencing Method Kit and the ABI PRISM 310 Bioanalyzer. For deleterious errors, OE-PCR can be utilized for correction [see Figure 6-6(a) for an example of the PULA gene]. Errors in the PAS end-products can be attributed to two main causes: errors in chemical synthesis of the oligonucleotides and errors in the product from PCR. Current methods used to synthesize oligonucleotides, especially when utilizing multi-frequency synthesizers for high-throughput synthesis of long oligonucleotides, often result in premature sequence termination or internal deletions. If unpurified oligonucleotides are used, the error rate will be as high as 5-10 errors per 1,000 bp synthesized. With PAGE-purified oligonucleotides, the error rate is reduced to about 1 error per 1,000 bp synthesized. Therefore, PAGE purification of oligonucleotides is essential. Although PAGE purification can remove oligonucleotides of incorrect length, it cannot detect and remove base substitution errors.
III. OE-PCR ValidationTo be able to correct each error, design an accurate pair of complementary primers in the reaction protocol. For example, in the synthesis of the PULA gene, three errors were found: A 589 G, C 1320 T, and T 2120 A [Figure 6-6(a)]. Therefore, six new oligonucleotides (25 bp in length) with the correct DNA sequence were chemically synthesized to cover the three erroneous parts. These six oligonucleotides are named PULA-M1, PULA-M2, PULA-M3, PULA-M4, PULA-M5, and PULA-M6 [Figure 6-6(b)]. Each pair of oligonucleotides covers an error region. For example, oligonucleotides PULA-M1 and PULA-M2 cover the region containing error A 589 G [Figure 6-6(a)]. Briefly, the first step of OE-PCR uses PCR-synthesized DNA fragments I, II, III, and IV [Fig. 6-6(a)], using the PULA DNA sequence produced by the PAS method as a template.DNA fragments I, II, III, and IV should have been corrected for three errors. The second step is to synthesize the complete PULA gene by PCR using the mixed DNA fragments I, II, III and IV as templates.

16. Number the PCR tubes and add 100 ng of full-length DNA from the second PCR synthesis to each tube. add 1 μL (30 pmol) of the appropriate upstream and downstream primers to the appropriate tube. For example, for PULA synthesis, for tube I, add PULA-1 and PU-LA-M2 [Figure 6-6(b)] to amplify DNA fragment I [Figure 6-6(a)]. For tube II, PULA-M1 and PULA-M4 [Figure 6-6(b)] were added to amplify DNA fragment II [Figure 6-6(a)]. For Tube III, PULA-M3 and PULA-M6 [Figure 6-6(b)] were added to amplify DNA fragment III [Figure 6-6(a)]. For tube IV, PULA-M5 and PULA-71 [Figure 6-6(b)] were added to amplify DNA fragment IV [Figure 6-6(a)].
The conditions for PCR are as follows:

17. Purify the PCR product by PAGE. 18.
18. Mix the PAGE-purified DNA fragments (100 ng each). 1 μL of the outermost oligonucleotide of each pair from step 10 was used as a primer to synthesize full-length DNA (GeneAmp PCR System 9 600). Perform PCR under the following conditions:

19. All PCR products were subjected to agarose gel electrophoresis on a 1.0% agarose gel at 150 V for 20 min to see if a single band was obtained as expected, followed by gene sequencing.
For more product details, please visit Aladdin Scientific website.